Click2Drug
Directory of computer-aided Drug Design tools
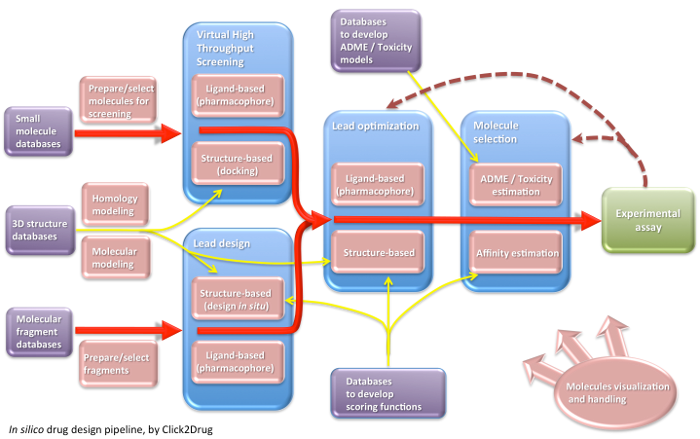
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services.These tools are classified according to their application field, trying to cover the whole drug design pipeline.
If you think that an interesting tool is missing in this list, please contact us.
Click on the following picture to select tools related to a given activity:
Show all links Hide all links
Databases
ZincDatabase, Zinc15Database, ChEMBL, Bingo, JChemforExcel, ChemDiff, ProteinDataBank(PDB), BindingMOAD(MotherOfAllDatabase), PDBbind, TTD, STITCH, SMPDB, ...
Chemical databases
- Zinc Database. Curated collection of commercially available chemical compounds, with 3D coordinates, provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Zinc15 Database. A new version of ZINC database including 100+ million purchasable compounds in ready-to-dock, 3D formats, provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- ChEMBL. Curated database of small molecules. Includes interactions and functional effects of small molecules binding to their macromolecular targets, and series of drug discovery databases.
- Chemspider. Collection of chemical compunds maintained by the Royal Society of Chemistry. Includes the conversion of chemical names to chemical structures, the generation of SMILES and InChI strings as well as the prediction of many physicochemical parameters.
- CoCoCo. Free suite of multiconformational molecular databases for High-Throughput Virtual Screening. It has single and multi conformer databases prepared for HTVS in different formats like Phase, Catalyst, Unity and SDF. Provided by the Department of Pharmaceutical Sciences of the University of Modena and Reggio Emilia.
- DrugBank. Bioinformatics and cheminformatics resource combining detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. Allows searching for similar compounds.
- PubChem. Database of chemical compounds maintained by the National Center for Biotechnology Information (NCBI), along with bioassays results. Allows similar compounds search (2D and 3D).
- TCM. Free small molecular database on traditional Chinese medicine, for virtual screening. It is currently the world's largest TCM database, and contains 170'000 compounds, with 3D mol2 and 2D cdx files, which passed ADMET filters.
- SCUBIDOO. a freely accessible database concept that currently holds 21 million virtual products originating from a small library of building blocks and a collection of robust organic reactions. This large data set was reduced to three representative and computationally tractable samples denoted as S, M, and L, containing 9994, 99 977, and 999 794 products, respectively. These small sets are useful as starting points for ligand identification and optimization projects. Proposed by the University of University of Marburg, Germany.
- Mcule database. Commercial database of commercially available small molecules. Allows filtering by chemical supplier data (stock availability, price, delivery time, chemical suppliers, catalogs, minimum purity, etc.) and export the whole Mcule database including supplier and procurement related properties. Reduced prices for academic. Provided by Mcule.
- WOMBAT. (World of Molecular Bioactivity). Database of 331,872 entries (268,246 unique SMILES), representing 1,966 unique targets, with bioactivity annotations. Compiled by Sunset Molecular Discovery LLC.
- Approved Drugs. The Approved Drugs app contains over a thousand chemical structures and names of small molecule drugs approved by the US Food & Drug Administration (FDA). Structures and names can be browsed in a list, searched by name, filtered by structural features, and ranked by similarity to a user-drawn structure. The detail view allows viewing of a 3D conformation as well as tautomers. Structures can be exported in a variety of ways, e.g. email, twitter, clipboard. For iPad and iPhone. Developed by Molecular Materials Informatics, Inc.
- ChemSpider Mobile. Allows searching the ChemSpider chemical database, provided by the Royal Society of Chemistry. Compounds can be searched by structure or by name, and browsed within the app. Results can be examined by jumping to the web page. Search structures are drawn using the powerful MMDS molecular diagram editor. For iPad. Provided by Molecular Materials Informatics, Inc.
- e-Drug3D. Database mirroring the current content of the U.S. pharmacopeia of small drugs. Contains 1822 molecular structures with a molecular weight < 2000 (last update: July 2016). Provides SD files (single conformer, tautomers or multiple conformers). Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.
- GLASS. GLASS (GPCR-Ligand Association) database is a manually curated repository for experimentally-validated GPCR-ligand interactions. Along with relevant GPCR and chemical information, GPCR-ligand association data are extracted and integrated into GLASS from literature and public databases. A list of currently-known GPCRs was compiled from UniProt and used to filter through the other chemical databases for ligand-association data (ChEMBL, BindingDB, IUPHAR, DrugBank, PDSP), GPCR diseases association (TTD), GPCR experimental structural data (PDB, BioLiP), and predicted models of GPCRs (GPCRRD). Subsequently, information from the extracted databases were unified to the same format and checked to ensure that all entries are only GPCR-related. Thus, the user would not find any entries on receptor tyrosine kinases or any other protein that is not a GPCR. All relevant ligand chemical data (PubChem) and GPCR data (UniProt) were extracted accordingly for each GPCR-ligand entry. Each molecule with a unique InChI key was considered a unique ligand entry in the database. Developed and maintained by the Zhang Lab at the University of Michigan, USA.
- ChemDB/ChemicalSearch. Find chemicals by various search criteria.
- Structural Database (CSD). Repository for small molecule crystal structures in CIF format. The CSD is compiled and maintained by the Cambridge Crystallographic Data Centre
- SPRESIweb. Integrated database containing over 8.7 million molecules, 4.1 million reactions, 658,000 references and 164,000 patents covering the years 1974 - 2009. Developed by InfoChem.
- MMsINC. Database of non-redundant, annotated and biomedically relevant chemical structures. Includes the analysis of chemical properties, such as ionization and tautomerization processes, and the in silico prediction of 24 important molecular properties in the biochemical profile of each structure. MMsINC supports various types of queries, including substructure queries and the novel 'molecular scissoring' query. MMsINC is interfaced with other primary data collectors, such as PubChem, Protein Data Bank (PDB), the Food and Drug Administration database of approved drugs and ZINC. provided by the CRS4 - Bioinformatics Laboratory, Parco Sardegna Ricerche, Italy.
- ZINClick. ZINClick is a database of triazoles generated using existing alkynes and azides, synthesizable in no more than three synthetic steps from commercially available products. This resulted in a combinatorial database of over 16 million of 1,4-disubstituted-1,2,3-triazoles (Molecular Weight < 1000), each of which is easily synthesizable, but at the same time new and patentable. Provided by the Università degli Studi del Piemonte Orientale "A. Avogadro".
- SPRESImobile. iPod, iPhone and iPad application providing direct access to ChemReact, a subset of the SPRESI structure and reaction database, which contains more than 400,000 unique reaction types and the related references. Developed by InfoChem.
- MORE. (MObile REagents). Mobile app, for iphone, ipad and android, which gives access to over 9 million molecules and 16 million chemical product variations offered by 56 different suppliers. Can search reagents by name, formula or by drawing a chemical structure. It is possible to limit the search to specific suppliers, bookmark the search results, and export small sdfiles. Allows converting a picture of a chemical structure taken from the iPhone camera into a structurally searchable molecule using OSRA (Optical Structure Recognition Application).
- KKB. (Kinase Knowledgebase). Database of kinase structure-activity and chemical synthesis data. Developed and maintained by Eidogen-Sertanty, Inc.
- iKinase Universal. iPad/iPhone application providing sample structure activity data from Eidogen-Sertanty's Kinase Knowledgebase (KKB). Exists in a Pro version (iKinasePro).
- DUD.E. (Database of Useful Decoys: Enhanced). DUD-E is designed to help test docking algorithms by providing challenging decoys. It contains a total 22,886 active compounds and their affinities against 102 targets, an average of 224 ligands per target. Also includes 50 decoys for each active, having similar physico-chemical properties but dissimilar 2-D topology. DUD-E is provided freely by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- DUD. (Directory of Useful Decoys). DUD is designed to help test docking algorithms by providing challenging decoys. It contains a total of 2,950 active compounds against a total of 40 targets. For each active, 36 "decoys" with similar physical properties (e.g. molecular weight, calculated LogP) but dissimilar topology. DUD is provided freely by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- GPCR-Bench. GPCR-Bench provides a high quality GPCR docking benchmarking set: 25 PDB structures covering all NR structures as of January 2015, and active and decoy compounds in the spirit of DUD. Provided by Heptares Therapeutics Ltd., UK.
- MUV. Maximum Unbiased Validation Datasets for Virtual Screening, with non-clumpy, spatially random topology. Provided by Carolo-Wilhelmina University.
- GLL. (GPCR Ligand Library). Database of 25145 ligands for 147 GPCRs. Associated with the GDD (GPCR Decoy Database). Provided by the Claudio N. Cavasotto Lab. of the Instituto de Biomedicina de Buenos Aires - Max Planck Society Partner (IBioBA-MPSP).
- GDD. (GPCR Decoy Database). For each ligand in GLL, 39 decoys were drawn from ZINC ensuring physical similarity of six properties (molecular weight, formal charge, hydrogen bond donors and acceptors, rotatable bonds and logP), but structural dissimilarity. Provided by the Claudio N. Cavasotto Lab. of the Instituto de Biomedicina de Buenos Aires - Max Planck Society Partner (IBioBA-MPSP).
- VDS. Virtual Decoy Sets for Molecular Docking Benchmarks. Similar to DUD but ignoring synthetic feasibility. Expected to be less biased with respect to physical similarity.
- LEADS-PEP. A benchmark dataset for assessing peptide docking performance. The set includes 53 protein-peptide complexes with peptide ranging from 3 to 12 residues. Several well-known small molecule docking program were tested. Provided by the Fraunhofer Institute for Molecular Biology and Applied Ecology, Germany.
- DNP. (Dictionary of Natural Products). Comprehensive and fully-edited database on natural products, arising from the Dictionary of Organic Compounds (DOC).The compilation of DNP is undertaken by a team of academics and freelancers who work closely with the in-house editorial staff at Chapman & Hall. Each contributor specialises in a particular natural product class (e.g. alkaloids) and reorganises and classifies the data in the light of new research so as to present it in the most consistent and logical manner possible.
- ChemIDPlus. Database of compounds and structures by US National Library of Medicine
- ChemBank. Public, web-based informatics environment created by the Broad Institute's Chemical Biology Program. Includes freely available data derived from small molecules and small-molecule screens, and resources for studying the data.
- eMolecules. Database of unique molecules from commercial suppliers
- GLIDA. GPCR-Ligand Database. Provides information on both GPCRs and their known ligands. Enterable either by GPCR search or ligand search. Maintained by the PharmacoInformatics Laboratory, Kyoto University.
- Comparative Toxicogenomics Database (CTD). Database of manually curated data describing cross-species chemical-gene/protein interactions and chemical and gene disease relationships to illuminate molecular mechanisms underlying variable susceptibility and environmentally influenced diseases.
- SuperDRUG2. Database of more than 4,600 active pharmaceutical ingredients. Annotations include drugs with regulatory details, chemical structures (2D and 3D), dosage, biological targets, physicochemical properties, external identifiers, side-effects and pharmacokinetic data. Different search mechanisms allow navigation through the chemical space of approved drugs. A 2D chemical structure search is provided in addition to a 3D superposition feature that superposes a drug with ligands already known to be found in the experimentally determined protein-ligand complexes. It has been added simulations of "physiologically-based" pharmacokinetics of drugs. The interaction check feature identifies potential drug-drug interactions and also provides alternative recommendations for elderly patients. Maintained by the University of Charité, Berlin, Germany.
- Ligand Expo. Formerly Ligand Depot. Provides chemical and structural information about small molecules within the structure entries of the Protein Data Bank.
- Glide Ligand Decoys Set. Collection created by selecting 1000 ligands from a one million compound library that were chosen to exhibit "drug-like" properties. Used in Glide enrichment studies. Provided by Schrödinger.
- Glide Fragment Library. Set of 441 unique small fragments (1-7 ionization/tautomer variants; 6-37 atoms; MW range 32-226) derived from molecules in the medicinal chemistry literature. The set includes a total of 667 fragments with accessible low energy ionization and tautomeric states and metal and state penalties for each compound from Epik. These can be used for fragment docking, core hopping, lead optimization, de novo design, etc. Provided by Schrödinger.
- Virtual library Repository. Libraries of 30,184 (redundant) and 4,544 small-molecule fragments, all less than 150 daltons in weight, derived from FDA-approved compounds using the python script fragmentizer. Distributed by the National Biomedical Computation Resource.
- NRDBSM. (Non Redundant Database of Small Molecules) is a database aimed specifically at virtual high throughput screening of small molecules. It has been developed giving special consideration to physicochemical properties and Lipinski's rule of five. Provided by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- Ligand Expo. Ligand Expo (formerly Ligand Depot) provides chemical and structural information about small molecules within the structure entries of the Protein Data Bank. Tools are provided to search the PDB dictionary for chemical components, to identify structure entries containing particular small molecules, and to download the 3D structures of the small molecule components in the PDB entry. A sketch tool is also provided for building new chemical definitions from reported PDB chemical components.
- ChEBI. (Chemical Entities of Biological Interest). Freely available dictionary of molecular entities focused on ‘small’ chemical compounds. provided by the European Bioinformatics Institute.
- KEGG DRUG. Comprehensive drug information resource for approved drugs in Japan, USA, and Europe unified based on the chemical structures and/or the chemical components, and associated with target, metabolizing enzyme, and other molecular interaction network information. Provided by the Kyoto Encyclopedia of Genes and Genomes.
Databases handling
- Bingo. Relational database management system (RDBMS) data cartridge that provides fast, scalable, and efficient storage and searching solution for chemical information. Bingo integrates the chemistry into Oracle, Microsoft SQL Server and PostgreSQL databases. Its extensible indexing is designed to enable scientists to store, index, and search chemical moieties alongside numbers and text within one underlying relational database server. Free software. Distributed by GGA software.
- JChem for Excel. Integrates structure handling and visualizing capabilities within a Microsoft Excel environment. Structures are fully supported within spreadsheets and be can viewed, edited, searched, resized, ordered, managed. Provided by ChemAxon.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
- IXTAB. Xtab is a transversal compounds library management tool to create, import, explore and analyse databases. Provided by Mind The Byte.
Protein-ligand complexes databases
- Protein DataBank (PDB). Databank of experimentally-determined structures of proteins, nucleic acids, and complex assemblies.
- Binding MOAD (Mother Of All Database). Subset of the Protein Data Bank (PDB), containing a collection of well resolved protein crystal structures with clearly identified biologically relevant ligands annotated with experimentally determined binding data extracted from literature. Maintained by the university of Michigan.
- PDBbind. Collection of experimentally measured binding affinity data (Kd, Ki, and IC50) exclusively for the protein-ligand complexes available in the Protein Data Bank (PDB). All of the binding affinity data compiled in this database are cited from original references.
- ProPairs. A Data Set for Protein-Protein Docking that dentifies and presents protein docking complexes and their unbound structures. They can be used as benchmark sets to develop or to test docking algorithms. Hosted by Macromolecular Modelling Group, Freie Universität Berlin, Germany.
- NRLiSt. (Nuclear Receptors Ligands and Structures Benchmarking DataBase). Non-commercial manually curated benchmarking database dedicated to the Nuclear Receptor(NR) ligands and structures pharmacological profiles. Provided by the Conservatoire National des Arts et Métiers - Paris.
- CCDC/Astex Validation set. The new CCDC/Astex test set consists of 305 protein-ligand complexes. All protonation states have been assigned by manual inspection. It is an extended version of the original GOLD validation test set.
- AffinDB. Freely accessible database of affinities for protein-ligand complexes from the PDB.
- Protein Ligand Database (PLD). Collection of protein ligand complexes extracted fom the PDB along with biomolecular data, including binding energies, Tanimoto ligand similarity scores and protein sequence similarities of protein-ligand complexes. Maintained by the University of Cambridge.
- BindingDB. Public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules.
- Ki Database. Provides information on the abilities of drugs to interact with an expanding number of molecular targets. The Ki database serves as a data warehouse for published and internally-derived Ki, or affinity, values for a large number of drugs and drug candidates at an expanding number of G-protein coupled receptors, ion channels, transporters and enzymes. Currently 55472 Ki values. Maintained by the NIMH Psychoactive Drug Screening Program.
- SCORPIO. Free online repository of protein-ligand complexes which have been structurally resolved and thermodynamically characterised.
- PDSP. Psychoactive Drug Screening Program. Provides screening of novel psychoactive compounds for pharmacological and functional activity at cloned human or rodent CNS receptors, channels, and transporters. Assays, Ki,...
- BAPPL complexes set. 161 protein-ligand complexes with experimental and estimated binding free energies calculated with the BAPPL server.
- DNA Drug complex dataset. Dataset of DNA-drug complexes consisting of 16 minimized crystal structures and 34 model-built structures, along with experimental affinities, used to validate PreDDICTA.
- Binding Database. Public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules. Maintained by the Center for Advanced Research in Biotechnology, University of Maryland Biotechnology Institute.
- Kuntz Protein Test Set. Set of 114 crystallographically determined protein-ligand structures used to test the docking program DOCK. Maintained by UCSF.
Target databases
- TTD. (Therapeutic Target Database). Database to provide information about the known and explored therapeutic protein and nucleic acid targets, the targeted disease, pathway information and the corresponding drugs directed at each of these targets. Also included in this database are links to relevant databases containing information about target function, sequence, 3D structure, ligand binding properties, enzyme nomenclature and drug structure, therapeutic class, clinical development status. All information provided are fully referenced.
Pathway databases
- STITCH. Resource to explore known and predicted interactions of chemicals and proteins. Chemicals are linked to other chemicals and proteins by evidence derived from experiments, databases and the literature. STITCH contains interactions for between 300,000 small molecules and 2.6 million proteins from 1133 organisms. Provided by the Beyer group of the Biotechnology Center TU Dresden.
- SMPDB. (Small Molecule Pathway Database). Interactive, visual database containing more than 350 small molecule pathways found in humans. SMPDB is designed specifically to support pathway elucidation and pathway discovery in metabolomics, transcriptomics, proteomics and systems biology. All SMPDB pathways include information on the relevant organs, subcellular compartments, protein cofactors, protein locations, metabolite locations, chemical structures and protein quaternary structures. Provided by the Departments of Computing Science & Biological Sciences, University of Alberta.
Chemical structure representations
ChemDraw, MarvinSketch, ACD/ChemSketch, jsMolEditor, Marvinmoleculeeditorandviewer, Ketcher, UCSFChimera, Pymol, OpenStructure, DaylightSMILES, InChI, TriposMol2, OpenBabel, Corina, Indigo, PoseView, PLiP, Ligplot+, E-Babel, Corinaonlinedemo, ChemicalIdentifierResolver, COSMOS, VEGAWE, PDBHydrogenAddition, DG-AMMOS, ChemMobi, ChemSpotlight, ...
2D drawing
- ChemDraw. Molecule editor developed by the cheminformatics company CambridgeSoft. For Windows and Mac.
- MarvinSketch. Advanced chemical editor for drawing chemical structures, queries and reactions developed by ChemAxon. Exists as an applet.
- ACD/ChemSketch. Molecule editor developed by ACD/Labs. Also available as freeware, with tools for 2D structure cleaning, 3D optimization and viewing, InChI generation and conversion, drawing of polymers, organometallics, and Markush structures. For Windows only.
- DataWarrior. Free Cheminformatics Program for Data Visualization and Analysis. DataWarrior combines dynamic graphical views and interactive row filtering with chemical intelligence. Scatter plots, box plots, bar charts and pie charts not only visualize numerical or category data, but also show trends of multiple scaffolds or compound substitution patterns. Compounds can be clustered and diverse subsets can be picked. Calculated compound similarities can be used for multidimensional scaling methods, e.g. Kohonen nets. Physicochemical properties can be calculated, structure activity relationship tables can be created and activity cliffs be visualized.
- JKluster. Tool of JChem for clustering, diversity calculations, and library comparisons based on molecular fingerprints and other descriptors. Useful in combinatorial chemistry, drug design, or other areas where a large number of compounds need to be analyzed. Provided by ChemAxon.
- SMARTSeditor. Graphic editing tool for generic chemical patterns. Based on the SMARTS language, chemical patterns can be created and edited interactively, similar to molecule editing in a chemical structure editor. The visualization of patterns is based on the visualization concept of the SMARTSviewer.Freely available for linux systems with 32 and 64 bit, windows 32bit systems and MacOS. Developed by the University of Hamburg.
- VLifeBase. Provides features to build a molecule from scratch using 2D Draw and conversion to 3D. The 3D editor allows addition, modification, replacement and deletion of atoms, bonds and groups, with Undo and Redo operations. Provided by VLife.
- ISIS/Draw. Chemical structure drawing program for Windows, published by MDL Information Systems. Free of charge for academic and personal use.
- ChemDoodle. Chemical structure environment with a main focus on 2D graphics and publishing to create media for structures, reactions and spectra. For Windows, Mac and Linux.
- ChemDraw for iPad. iPad application to create, edit and share publication-quality chemical structures with just the touch of a finger, based on the world’s most popular chemical drawing software, ChemDraw. Provided by PerkinElmer, Inc.
- TouchMol Deskop Application. Tool for drawing chemical and biological structures, optimized for Touch Operations. Allows Copy/Paste to ChemDraw, ISIS/Draw, SciFinder and Word. Provides name-to-structure. For Windows 8. Provided by Scilligence.
- TouchMol for Office. Desktop tool for drawing chemical and biological structures, into the MS Office suite. Provided by Scilligence.
- ChemDoodle Mobile. Free iPhone companion to ChemDoodle. ChemDoodle Mobile is a calculator for drawn organic structures. There are four main windows: Draw, Calculate, Spectra and Help. The Draw window shows a typical ChemDoodle sketcher, where you can draw and store your structures. The Calculate page calculates properties and the Spectra page simulates NMR spectra. All spectra are interactive. The Help page contains a thorough help guide. Provided by iChemLabs.
- Chirys Draw. Application for drawing publication-quality molecular structures and reactions. Designed from the ground up for the iPad. Developed by Integrated Chemistry Design, Inc.
- Chirys Sketch. Application for drawing publication-quality molecular structures and reactions, for iPhone and iPod Touch. Developed by Integrated Chemistry Design, Inc.
- Mobile Molecular DataSheet. Allows viewing and editing chemical structure diagrams on an iPhone, iPod or iPad. Molecules are organized in collections of datasheets. Individual molecules, or whole datasheets, can be shared via iTunes or sent by email, using the standard MDL MOL and SDfile formats, which allows the data to be integrated into any external workflow. Provided by Molecular Materials Informatics, Inc.
- SAR Table. Application designed for creating tables containing a series of related structures, their activity/property data, and associated text. Structures are represented by scaffolds and substituents, which are combined together to automatically generate a construct molecule. The table editor has many convenience features and data checking cues to make the data entry process as efficient as possible. For iPad. Provided by Molecular Materials Informatics, Inc.
- Molprime+. Chemical structure drawing tool based on the unique sketcher from the Mobile Molecular DataSheet. Can send structure data via email, open structures from email or web, create graphical images or Microsoft Word documents with embedded structure graphics, calculate properties based on structures and use structures to search Mobile Reagents and ChemSpider. Provided by Molecular Materials Informatics, Inc.
- StructureMate. Portable chemical dataset viewer for iPad, for browsing SAR reports, chemical catalogs, custom-made databases, and physical property references. Provided by Metamolecular, LLC.
- Elemental. Chemistry sketch for iphone and ipad. Developed by Dotmatics Limited.
- Accelrys Draw. Allows drawing and editing complex molecules, chemical reactions and biological sequences. provided by Accelrys.
- PLT. Program for producing chemical drawings and outputting them in a variety of formats. For Windows.
- JChemPaint. Free and open source editor and viewer for chemical structures in 2D. Exists as a Java stand alone application and two varieties of Java applet that can be integrated into web pages. Platform-independent.
- BKchem. BKChem is a free open source chemical drawing program written in Python. Platform-independent.
- MolSketch. Free open source molecular drawing tool for 2D molecular structures. Available for Windows, Mac and Linux.
- JME Molecular Editor. Java applet which allows to draw / edit molecules and reactions (including generation of substructure queries) and to depict molecules directly within an HTML page. Editor can generate Daylight SMILES or MDL Molfile of created structures.
- Chem4D. Molecular drawing tool. Includes assignment of systematic names to organic structures according to IUPAC nomenclature rules, and drawing of molecules from IUPAC names. For Windows and Mac. Distributed by ChemInnovation Software.
- XDrawChem. Free open source software program for drawing chemical structural formulas, available for Windows, Unix, and Mac OS.
- iMolecular Draw. Application that can view, edit and build molecules in 2D. For iPhone.
- SketchEl. Free and open source interactive chemical molecule sketching tool, and molecular spreadsheet data entry application. Written in Java. Exists as an applet.
- Chemtool. Free open source program for drawing chemical structures on Linux and Unix systems using the GTK toolkit under X11.
- Bioclipse. Java-based, open source, visual platform for chemo- and bioinformatics based on the Eclipse Rich Client Platform (RCP).
- Chrawler. Can scan all data sources, including local files, remote files on network, emails, web pages, SharePoint contents, etc., and find contained chemical structures, and make them structure-searchable (substructure, full-structure, similarity). Distributed by Scilligence.
- Imago. Toolkit for 2D chemical structure image recognition. It contains a GUI program and a command-line utility, as well as a documented API for developers. Imago is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- Imago OCR Visual Tool. Java GUI for Imago. Ego is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- Imago Console Application. Command-line interface for Imago. Alter-Ego is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- OLN Chem4SharePoint. Makes it possible to draw, display and search chemical structures in SharePoint. Distributed by Scilligence.
- ChemJuice. Molecular drawing software for iPhone. Developed by IDBS.
- ChemJuice Grande. Molecular drawing software for iPad. Developed by IDBS.
- MolPad. Free chemical structure drawing application. It can draw structures from scratch or load them from ChemSpider and modify them. Structures can be emailed in Molfile format. For Android.
- DCE ChemPad. Free application to draw chemical structures and calculate molecular weight, molecular formula and to send the molfile. It shows the capabilities of the Dendro Chemical Editor control for Android to build chemistry-aware mobile applications. For Android.
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
2D drawing online
- jsMolEditor. Molecule Editor in JavaScript. Open source.
- Marvin molecule editor and viewer. Java based chemical editor for drawing chemical structures. Includes unlimited structure based predictions for a range of properties (pKa, logD, name<>structure, etc.). Provided by ChemAxon.
- Ketcher. Web-based chemical structure editor written in JavaScript. Free and open-source, but also available on a commercial basis. Distributed by GGA software.
- ChemWriter. Chemical structure editor designed for use with Web applications. Distributed by Metamolecular.
- Molinspiration WebME Molecule Editor. Allows creation and editing of molecules in browsers without Java support and without any plugins. The editor is based on a Web2.0 Ajax technology. WebME allows therefore web-based structure input also in institutions where Java applets are not allowed and offers complete platform compatibility. The actual molecule processing in WebME is based on the JMEPro editing engine running on a server. provided by Molinspiration.
- OLN JSDraw. Javascript libary you can display and draw chemical structures in web pages, which works cross browser, including IE, Firefox, Safari, Opera and Chrome, crose platform, including Window, Mac, Linux, and even iPhone, Android and other mobile devices. Free for education. Provided by Scilligence.
- TouchMol Web. Tool for drawing chemical and biological structure online. Allows Copy/Paste to ChemDraw, ISIS/Draw, SciFinder and Word. Provides name-to-structure. Provided by Scilligence.
3D viewers
- UCSF Chimera. Open source, highly extensible program for interactive visualization and analysis of molecular structures and related data. Free of charge for academic, government, non-profit, and personal use. For Windows, Mac and Linux. Developed by the Resource for Biocomputing, Visualization, and Informatics, UCSF.
- Pymol. Open source, user-sponsored, molecular visualization system written in Python. Distributed by DeLano Scientific LLC. For Windows, Mac and Linux.
- OpenStructure. Open-source, modular, flexible, molecular modelling and visualization environment. It is targeted at interested method developers in the field of structural bioinformatics. Provided by the Swiss Institute of Bioinformatics and the Biozentrum, University of Basel.
- Swiss-PDB Viewer / DeepView. Program for 3D visualization of macromolecules, allowing to analyze several proteins at the same time. Swiss-PdbViewer is tightly linked to SWISS-MODEL, an automated homology modeling server developed within the Swiss Institute of Bioinformatics (SIB).
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- Computer-Aided Drug-Design Platform using PyMOL. a simple Java tool for visual exploration of three-dimensional (3D) virtual screening data. The VSviewer3D brings together the ability to explore numerical data, such as calculated properties and virtual screening scores, structure depiction, interactive topological and 3D similarity searching, and 3D visualization. By doing so the user is better able to quickly identify outliers, assess tractability of large numbers of compounds, visualize hits of interest, annotate hits, and mix and match interesting scaffolds. We demonstrate the utility of the VSviewer3D by describing a use case in a docking based virtual screen. Developed by Data2Discovery Consulting Inc., USA.
- Autodock Vina plugin for PyMOL. Allows defining binding sites and export to Autodock and VINA input files, doing receptor and ligand preparation automatically, starting docking runs with Autodock or VINA from within the plugin, viewing grid maps generated by autogrid in PyMOL, handling multiple ligands and set up virtual screenings, and set up docking runs with flexible sidechains.
- Dehydron. A dehydron calculator plugin for PyMOL. This plugin calculates dehydrons and display them onto the protein structure.
- pymacs. Python module for dealing with structure files and trajectory data from the GROMACS molecular dynamics package. It has interfaces to some gromacs functions and uses gromacs routines for command line parsing, reading and writing of structure files (pdb,gro,...) and for reading trajectory data (only xtc at the moment).
- PyRosetta. Interactive Python-based interface to the Rosetta molecular modeling suite. It enables users to design their own custom molecular modeling algorithms using Rosetta sampling methods and energy functions.
- Visual Molecular Dynamics (VMD). Free open source molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting. For MacOS X, Unix, or Windows. Developed by the NIH resource for macromolecular modeling and bioinformatics, University of illinois.
- ePMV. (embedded Python Molecular Viewer). Free, open-source plug-in that runs molecular modeling software directly inside of professional 3D animation applications (hosts, i.e. Blender, Cinema4D and Maya 2011) to provide simultaneous access the capabilities of all of the systems. Developed by the Scripps Research Institute.
- Jmol. Open source Java viewer for chemical structures in 3D.
- Zodiac. Free open source molecular modelling suite for computation, analysis and display of molecular data. It features state-of-the-art tools for managing molecular databases, run molecular docking experiments, compute raytraced images, etc... Developed by Zeden. For windows, Mac and Linux.
- GLmol. Free and open source 3D molecular viewer based on WebGL and Javascript. GLmol runs on newer versions of Firefox, Chrome, Safari or Opera. Internet Explorer is not supported. GLmol also runs on Sony Ericsson's Android devices which support WebGL and WebGL enabled safari in iOS.
- DS Visualizer. Free 3D visualizer of Discovery Studio. Allows sequence handling and, 2D or 3D charting. Creates 2D ligand-receptor interaction diagrams. Distributed by Accelrys. DS Visualizer ActiveX Control allows visualizing and interacting with molecules in Microsoft Office documents and Internet Explorer. For Windows and Linux.
- OpenAstexViewer. Free open source java molecular graphics program that assists in structure based drug design. It can be used as an Applet in a web page or as a desktop application. Provided by Astex Therapeutics. For Windows, linux and Mac.
- ICM-Browser. Free molecular visualization program for displaying proteins, DNA and RNA, and multiple sequence alignments. Allows saving interactive 3D files to display on the web or in PowerPoint. Distributed by Molsoft. For Windows, Mac and linux. Exist in a Pro version.
- Crystal Studio. Crystal Studio is a Windows XP/Vista/Windows 7 (32/64) software package for crystallography. It is a comprehensive tool for user-friendly creation, 3D graphical design, display and manipulation of crystal and macro-molecular structures, surface or interfaces and defects and for the simulation of X-Ray, neutron and electron diffraction patterns.
- Friend. Integrated Front-End application for multiple structure visualization and multiple sequence alignment. Friend is a bioinformatics application designed for simultaneous analysis and visualization of multiple structures and sequences of proteins and/or DNA/RNA. The application provides basic functionalities such as: structure visualization with different rendering and coloring, sequence alignment, and simple phylogeny analysis, along with a number of extended features to perform more complex analyses of sequence structure relationships, including: structure alignment of proteins, investigation of specific interaction motifs, studies of protein-protein and protein-DNA interactions, and protein super-families. Friend is also available as an applet. Provided by the Ray and Stephanie Lane Center for Computational Biology.
- Chemkit. Free open-source C++ library for molecular modelling, cheminformatics, and molecular visualization.
- Coot. Program for macromolecular model building, model completion and validation, particularly suitable for protein modelling using X-ray data. Free and open-source.
- Jamberoo. Free open source program for displaying, analyzing, editing, converting, and animating molecular systems (former JMolEditor). For Windows, Mac and Linux.
- YASARA View. Free molecular visualization program for displaying macromolecules, building molecules, multiple sequence alignments. Can be complemented by YASARA Model. Provided by YASARA.
- QuteMol. Open source (GPL), interactive, high quality molecular visualization system. QuteMol exploits the current GPU capabilites through OpenGL shaders to offers an array of innovative visual effects. QuteMol visualization techniques are aimed at improving clarity and an easier understanding of the 3D shape and structure of large molecules or complex proteins. Developed by the Visual Computing Lab at ISTI-CNR, Italy.
- Molekel. Free open-source multi-platform molecular visualization program, for Mac OSX, Windows and Linux. Provided by the Swiss National Supercomputing Centre: Lugano (Switzerland).
- NOC. Free molecular explorer for protein structure visualization, validation and analysis. Mainained by Dr. Nymeyer's Group, Inst. Mol. Biol., Florida State University.
- CueMol. Program for the macromolecular structure visualization (CueMol was formerly called "Que"). CueMol aims to visualize the crystallographic models of macromolecules with the user-friendly interfaces. Currently supported files are molecular coordinates (PDB format), electron density (CCP4, CNS , and BRIX formats), MSMS surface data, and APBS electrostatic potential map.
- TexMol. Molecular visualization and computation package. Free and open source software.
- Chil2 Viewer. Visualization tool and graphical user interface of the Chil2 suite, with analysis tools, database integration and ruby interface. Open for general research.
- VEGA ZZ. Visualization application and molecular modeling toolkit (Molecular mechanics and dynamics, structure-based screening). Free for non-profit academic uses. Provided by the Drug Design Laboratory of the University of Milano.
- BALLView. Standalone molecular modeling and visualization application. Provides a framework for developing molecular visualization functionality. Can be used as the visualizaion component of BALL. Free and opensource. For Windows, Mac and Linux.
- RasMol. Program for molecular graphics visualisation.
- RasTop. Free open source molecular visualization software adapted from the program RasMol. RasTop wraps a user-friendly graphical interface around the "RasMol molecular engine". Developed for educational purposes and for the analysis of macromolecules at the bench. For Windows and Linux.
- Cn3D. Visualization tool for biomolecular structures, sequences, and sequence alignments. Maintained and distributed by the NCBI. For Windows, Mac and Linux.
- Bodil. Free, modular, multi-platform software package for biomolecular visualization and modeling. Bodil aims to provide easy three-dimensional molecular graphics closely integrated with sequence viewing and sequence alignment editing.
- COSMOS Viewer. Free software for presentation of molecules.
- BARISTA. BARISTA visualization functions create, display, and manipulate 3D depictions of molecular structures based on results computed by molecular computation programs such as Conflex, and are designed specifically to facilitate the analysis of these results. For Windows and Linux.
- BioAdviser. Visualization tool for biomolecular structures and small molecules.
- iMolview. Application to browse and view in 3D protein and DNA structures from Protein Data Bank, and drug molecules from DrugBank For iPhone and iPad. Provided by Molsoft.
- PyMOL on the iPad.. High-performance 3D molecular visualizer, designed from the ground up for the iPad. it can search and download data from the PDB, PubChem, Dropbox, or an own secure custom PyMOL enterprise server. Provided by Schrödinger.
- RCSB PDB.. The RCSB Protein Data Bank (PDB) mobile app is the official mobile app of the RCSB PDB. It provides fast, on-the-go access to the RCSB PDB resources. The app enables the general public, researchers and scholars to search the Protein Data Bank and visualize protein structures using either a WiFi or cellular data connection.
- Ball&Stick. High-quality molecular visualization app for the iPad, iPhone and iPod Touch. Provided by MolySym.
- CueMol for iOS. Interactive macromolecular viewer for structural biologists. CueMol viewer allows the users to open and view the scene files made by the desktop version of CueMol, and the Protein Data Bank (PDB) format files, as well.
- 3D Molecules Edit&Drill. Application designed to enable students and professionals to build, construct, modify and examine molecules in 3D. Allows the users to open and view molecules in SDF format files, for example, from NCBI's PubChem. Developed by Virtualnye Prostranstva LLC.
- Chem3D for iPad. Chem3D for iPad enables scientists to view and manipulate 3D images of chemical and biochemical structures. Re-imagined for the iPad, the Chem3D app features a facile user interface to manipulate images using common touch, pinch and swipe gestures. Provided by PerkinElmer, Inc.
- CMol. Interactive 3D molecular viewer designed specifically for the iPad, iPhone and iPod touch. CMol allows the user to open and view PDB files with complete control over the representations and colours used for individual chains, residues and atoms.
- Molecules. Free application for iPhone and iPad, for viewing three-dimensional renderings of molecules and manipulating them using your fingers. You can rotate the molecules by moving your finger across the display, zoom in or out by using two-finger pinch gestures, or pan the molecule by moving two fingers across the screen at once. These structures can be viewed in both ball-and-stick and spacefilling visualization modes.
- iMolecular Builder. The IMoleBuilder is an application that can view, edit and build molecules in 3D. For iPhone.
- iPharosDreams. Molecular visualization app for iPad to perform in-silico drug discovery. Downloads protein structure files from Protein Data Bank, displays 3D molecules, touch, rotation, zoom in/out. Hierarchy structure of molecules is shown with a table that select components in a protein and related things. It can generate pharmacophores and analyze 3D protein-ligand interaction of biological macromolecules for in-silico drug discovery. Allows selecting a ligand from a protein and generate a binding site from the selected ligand. Can generate receptor based pharmacophores and get inspiration. Developed by EQUISnZAROO CO., LTD.
- Jmol Molecular Visualization. Free Jmol for Android tablets.
- NDKmol. Free molecular viewer for Android.
- Molecule Viewer 3D. Opens most common 3D molecule file formats saved on a SD card or found in a library of 243 included molecules. For Android.
- 3D Molecule View. 3D molecule viewer. For Android.
- Atomdroid. Free computational chemistry tool. It can be used as a molecular viewer/builder and contains local optimization and Monte Carlo simulation features. For Android.
- Atom 3D. Free application to visualize molecules and crystal structures in 3D using the touchscreen to rotate and zoom. Includes 19 sample structures. Supports XYZ files and some protein data bank (PDB) files. For Android.
- PDBs. Free application for molecular graphics visualization from PDB files. For Android.
- PDB View 3D. Application for molecular graphics visualization from PDB files. For Android.
Definitions and syntax of file formats
- Daylight SMILES. SMILES (Simplified Molecular Input Line Entry System) is a line notation (a typographical method using printable characters) for entering and representing molecules and reactions.
- InChI. (IUPAC International Chemical Identifier) is a string of characters capable of uniquely representing a chemical substance. It is derived from a structural representation of that substance in a way designed to be independent of the way that the structure was drawn (thus a single compound will always produce the same identifier). It provides a precise, robust, IUPAC approved tag for representing a chemical substance.
- Tripos Mol2. Complete description of the Mol2 file format (.mol2).
- PDB format. Complete description of the PDB file format (.pdb).
- SDF format. Complete description of the SDF file format (.sdf).
- SMARTS format. SMARTS Tutorial by Daylight.
- OpenSMILES. Community sponsored open-standards version of the SMILES language for chemistry. OpenSMILES is part of the Blue Obelisk community.
File format Converters
- OpenBabel. Free open source chemical expert system mainly used for converting chemical file formats. For Windows, Unix, and Mac OS.
- Corina. Generates 3D structures for small and medium sized, drug-like molecules. Distributed by Molecular Networks.
- Indigo. Universal organic chemistry toolkit, containing tools for end users, as well as a documented API for developers. Free and open-source, but also available on a commercial basis. Distributed by GGA software.
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
- Indigo-cano. Command-line canonical SMILES generator. Free and open source. Distibuted by GGA software.
- Indigo-deco. Command-line program for R-Group deconvolution. Free and open source. Distibuted by GGA software.
- OMEGA. (Conformer Ensembles Containing Bioactive Conformations). Converts from 1D or 2D to 3D using distance bounds methods, with a focus on reproducing the bioactive conformation. Developed by OpenEye.
- COSMOS. (COordinates of Small MOleculeS). High-throughput method to predict the 3D structure of small molecules from their 1D/2D representations. Also exists as a web service. Provided by the University of california, Irvine.
- TorsionAnalyzer. Generate and analyse 3D conformers of small molecules. TorsionAnalyzer is based on an expert-derived collection of SMARTS patterns and rules (assigned peaks and tolerances). Rules result from statistical analysis of histograms derived from small molecule X-ray data extracted from the CSD. Rotatable bonds of molecules loaded into the TorsionAnalyzer are color-coded on the fly by means of a traffic light highlighting regular, borderline and unusual torsion angles. This allows the user to see at a glance if one or more torsion angles are out of the ordinary. Provided by BioSolveIT.
- LigPrep. 2D to 3D structure conversions, including tautomeric, stereochemical, and ionization variations, as well as energy minimization and flexible filters to generate ligand libraries that are optimized for further computational analyses. Distributed by Schrodinger.
- CACTVS. Universal scriptable toolkit for chemical information processing. Used by PubChem. Maintained and distributed by Xemistry. Free for academic.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
- OSRA. (Optical Structure Recognition Application). Utility designed to convert graphical representations of chemical structures, as they appear in journal articles, patent documents, textbooks, trade magazines etc. OSRA can read a document in any of the over 90 graphical formats parseable by ImageMagick - including GIF, JPEG, PNG, TIFF, PDF, PS etc., and generate the SMILES or SDF representation of the molecular structure images encountered within that document. Free and open source. Developed by the Frederick National Laboratory for Cancer Research, NIH.
- MayaChemTools. Collection of Perl scripts, modules, and classes to support day-to-day computational chemistry needs. Free software, open source. Provided by Manish Sud.
- VLife Engine. Engine module of VLifeMDS containing basic molecular modeling capabilities such as building, viewing, editing, modifying, and optimizing small and arge molecules. Fast conformer generation by systematic and Monte-carlo methods. Provided by VLife.
- SPORES. (Structure PrOtonation and REcognition System). Structure recognition tool for automated protein and ligand preparation. SPORES generates connectivity, hybridisation, atom and bond types from the coordinates of the molecule`s heavy atoms and hydrogen atoms to the structure. The protonation can either be done by just adding missing hydrogen atoms or as a complete reprotonation. SPORES is able to generate different protonation states, tautomers and stereoisomers for a given structure. Developed by the Konstanz university.
- DG-AMMOS. Program to generate 3D conformation of small molecules using Distance Geometry and Automated Molecular Mechanics Optimization for in silico Screening. Freely distributed by the University of Paris Diderot.
- Key3D. Molecular modeling tool to convert 2D structures (chemical structural formula) of compounds drawn by ISIS-Draw or ChemDraw to 3D structures with additional information on atomic charge etc. Distributed by IMMD.
- ChemDoodle. A software suite for drawing chemical structure diagrams, including the ability to calculate NMR spectra, generate IUPAC names and line notations for structures, manipulate structures imported from the Internet, interpret and interconvert files generated by other chemical drawing software programs, illustrate glassware and equipment setups, and draw TLC plates. Distributed by iChemLabs LLC.
- CONFLEX. Software for searching and analyzing the conformational space of small and large molecules.
- JOElib. Cheminformatics library mainly used for conversion of file formats. Written in Java. For Windows, Unix, and Mac OS.
- CDK (Chemistry Development Kit). LGPL-ed library for bio- and cheminformatics and computational chemistry written in Java. Opensource.
- MolEngine. .NET Cheminformatics Toolkit completely built on Microsoft .NET platform. By using Mono, MolEngine can run on other platform, such as Mac, Linux, iPad. Distributed by Scilligence.
- Indigo. Universal organic chemistry toolkit. Free and opensource. Provided by GGA.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Provided by GGA.
- Open Drug Discovery Toolkit. ODDT is a free and open source tool for both computer aided drug discovery (CADD) developers and researchers. It reimplements many state-of-the-art methods, such as machine learning scoring functions (RF-Score and NNScore) and wraps other external software to ease the process of developing CADD pipelines. ODDT is an out-of-the-box solution designed to be easily customizable and extensible. Therefore, users are strongly encouraged to extend it and develop new methods. Provided by the Institute of Biochemistry and Biophysics PAS, Warsaw, Poland.
- RDKit. Collection of cheminformatics and machine-learning software written in C++ and Python.
- Mol2Mol. Molecule file manipulation and conversion program.
- Fconv. Molecule file manipulation and conversion program.
- Knodle. KNOwledge-Driven Ligand Extractor is a software library for the recognition of atomic types, their hybridization states and bond orders in the structures of small molecules. Its prediction model is based on nonlinear Support Vector Machines. The process of bond and atom properties perception is divided into several steps. At the beginning, only information about the coordinates and elements for each atom is available: (i) Connectivity is recognized; (ii) A search of rings is performed to find the Smallest Set of Smallest Rings (SSSR); (iii) Atomic hybridizations are predicted by the corresponding SVM model; (iv) Bond orders are predicted by the corresponding SVM model; (v) Aromatic cycles are found and (vi) Atomic types are set in obedience to the functional groups. Some bonds are reassigned during this stage. Linux and MacOS version are free of charge. Maintained by the Nano-D team, Inria/CNRS Grenoble, France.
- smi23d. Consists of two programs that can be used to convert one or more SMILES strings to 3D. For Mac and Linux. Also exists as a web service.
- Scaffold Hunter. JAVA-based software tool for exploring the chemical space by enabling generation of and navigation in a scaffold tree hierarchy annotated with various data. The graphical visualization of structural relationships allows to analyze large data sets, e.g., to correlate chemical structure and biochemical activity. Free open source software developed and supported by the Chair of algorithm Engineering at Technical University Dortmund and the Department of Chemical Biology at Max-Planck Institute for Molecular Physiology Dortmund.
- ScaffoldTreeGenerator. Java-based program which generates the scaffold tree database independently of Scaffold Hunter. Free open source software developed and supported by the Chair of algorithm Engineering at Technical University Dortmund and the Department of Chemical Biology at Max-Planck Institute for Molecular Physiology Dortmund.
- Strip-it. Program to extract scaffolds from organic drug-like molecules by 'stripping' away sidechains and representing the remaining structure in a condensed form. Open source software distributed by Silicos.
- fragmentizer. Free and open source python script that can decompose PDBs of small-molecule compounds into their constituent fragments. Developed by the National Biomedical Computation Resource.
- Epik. Enumerates ligand protonation states and tautomers in biological conditions. Distributed by Schrodinger.
- iBabel. iBabel is an alternative graphical interface to Open Babel for Macintosh OS X.
- PerlMol. Collection of perl modules providing objects and methods for representing molecules, atoms, and bonds in Perl; doing substructure matching; and reading and writing files in various formats.
- The SDF Toolkit in Perl 5. The purpose of this SDF toolkit is to provide functions to read and parse SDFs, filter, and add/remove properties.
Analysis of ligand-protein interactions
- PoseView. Automatically generates 2D structure-diagrams of protein-ligand complexes (png, svg and pdf) provided as 3D-input. Such input may come directly from crystal structures or be computed for example by a docking program. PoseView images are available for the majority of PDB-structures on the PDB web site. Developed by the University of Hamburg and distributed by BioSolveIT.
- PLiP. Web service and command line tool for fully automated characterization and analysis of non-covalent interactions between proteins and ligands in 3D structures. Developed by the Technische Universität of Desden, Germany.
- Ligplot+. Java interface of Ligplot, a program for automatic generation of 2D ligand-protein interaction diagrams. Developed and proposed free-for-non-profit by the European Bioinformatics Institute (EMBL-EBI).
- LeView. Java program that to generate 2D representations of ligands and their environments and binding interactions for PDB entries. It can be used automatically (in command line) or interactively (with a graphical interface). Provided free of charge by the Institut Pasteur de Lille, France.
- DS Visualizer. Free 3D visualizer of Discovery Studio. Allows sequence handling and, 2D or 3D charting. Creates 2D ligand-receptor interaction diagrams. Distributed by Accelrys. DS Visualizer ActiveX Control allows visualizing and interacting with molecules in Microsoft Office documents and Internet Explorer. For Windows and Linux.
- BINANA. (BINding ANAlyzer). Python-implemented algorithm for analyzing ligand binding. The program identifies key binding characteristics like hydrogen bonds, salt bridges, and pi interactions. As input, BINANA accepts receptor and ligand files in the PDBQT format. Allows visualization with VMD. Developed by the National Biomedical Computation Resource.
Web services
- E-Babel. Online version of OpenbBabel. Maintained by the Virtual Computational Chemistry Laboratory.
- Corina online demo. Online demo of CORINA. Generates 3D coordinates from SMILES.
- Chemical Identifier Resolver. Converts a given structure identifier into another representation or structure identifier, using CACTVS. May give the name of a given molecule from SMILES of InChi, thanks to a database of 68 million chemical names linked to 16 million unique structure records.
File format Converters
- COSMOS. (COordinates of Small MOleculeS). High-throughput method to predict the 3D structure of small molecules from their 1D/2D representations. Also exists as a standalone program. Provided by the University of california, Irvine.
Web services
- VEGA WE. Web server for file translation tool, properties and surface calculation. Provided by the Drug Design Laboratory of the University of Milano.
- PDB Hydrogen Addition. Tool to add the hydrogen in a given PDB (for protein, DNA and drugs).
- DG-AMMOS. Generates single 3D conformer for small compound.
- Frog2. FRee Online druG conformation generation.
- Smiles2Monomers. Smiles2Monomers is a software to infer monomeric structure of polymers from their atomic structure. The web server is available for peptide-like compounds in the second tab and provides an interface to upload a compound in the SMILES format to compute the monomeric structure in two different formats: text formats (the structure is downloadable in json and xml) or image format (the colored picture of the monomeric structure mapped on the atomic structure is directly available in the browser or downloadable into a zip file). Provided by the University of Lille, France.
- e-LEA3D. Draw a molecule by using the ACD applet (v.1.30) and generate 3D coordinates by using the program Frog.
- MolEdit. Web server for 2D molecular editor & 3D structure optimization. Provided by the Drug Design Laboratory of the University of Milano.
- Chemozart. Chemozart is a 3D Molecule editor and visualizer built on top of native web components. It offers an easy to access service, user-friendly graphical interface and modular design. It is a client centric web application which communicates with the server via a representational state transfer style web service. Both client-side and server-side application are written in JavaScript. A combination of JavaScript and HTML is used to draw three-dimensional structures of molecules. Provided by the Department of Chemistry, Shahid Beheshti University, Tehran, Iran
- ProBuilder. Protein/peptide builder from 1D to 3D. Provided by the Drug Design Laboratory of the University of Milano.
- Online SMILES Translator and Structure File Generator. Translates SMILES into SDF, PDB of MOL formats, possibly generating 3D coordinates.
- smi23D web service. Translates SMILES strings or a URL to a SMILES file and get back the 3D coordinates in SDF. users can get the SDF file by typing directly the SMILES in the web browser, e.g. http://rest.rguha.net/threed/d3.py/get3d?smiles=c1ccccc1
- iview. Interactive WebGL visualizer of protein-ligand complex. Developed by the Chinese university of Hong Kong.
- PoseView. Automatically generates 2D structure-diagrams of protein-ligand complexes provided as 3D-input. Such input may come directly from crystal structures or be computed for example by a docking program. Developed by the University of Hamburg and distributed by BioSolveIT.
- LCT. The Ligand Contact Tool calculates contacts between protein and ligand atoms, several parameters are available (distance cut-off, Van Der Waals radii usage, etc). Queries acepted are uploadable PDB format file or PDB accession code. Provided by the Structural Computational Biology Group of the Spanish national Cancer Research Centre.
- SimiCon. Identifies the equivalent protein-ligand atomic contacts between Reference and Target complexes. Results are shown as text, tables and 3D interactive graphics
- Smi2Depict. Webservice to generate 2D images from SMILES.
- GIF/PNG-Creator. GIF/PNG-Creator for 2D Plots of Chemical Structures from SMILES or structure files, using CACTVS. Maintained by the National Cancer institute, NIH.
- depict. Webservice using the molconvert tool of ChemAxon to generate 2D images from SMILES.
- SMARTSviewer. Webservice to visualize 2D images from SMARTS.
- OSRA web service. (Optical Structure Recognition Application). Web service designed to convert graphical representations of chemical structures, as they appear in journal articles, patent documents, textbooks, trade magazines etc. OSRA can read a document in any of the over 90 graphical formats parseable by ImageMagick - including GIF, JPEG, PNG, TIFF, PDF, PS etc., and generate the SMILES or SDF representation of the molecular structure images encountered within that document. Free and open source. Developed by the Frederick National Laboratory for Cancer Research, NIH.
Others
- ChemMobi. ChemMobi is a tool for Chemists, Biochemists and anyone else interested in chemical structures, chemical sourcing, chemical properties and safety information. For iPhone.
- ChemSpotlight. ChemSpotlight is a plugin for Mac OS X 10.5 and later, which reads common chemical formats and provides searching and preview in the Finder. ChemSpotlight reads common chemical file formats using the Open Babel chemistry library. Spotlight can then index and search chemical data: molecular weights, formulas, SMILES, InChI, fingerprints, etc. Developed by Geoffrey Hutchison. Free and open source.
Molecular Modeling
CHARMM, GROMACS, Amber, SwissParam, CHARMM-GUI, CHARMMing.org, ...
Software
- CHARMM. (Chemistry at HARvard Macromolecular Mechanics). Package of molecular simulation programs, including source code and demos.
- GROMACS. (GROningen MAchine for Chemical Simulations). Free open source molecular dynamics simulation package.
- Amber. (Assisted Model Building with Energy Refinement). Package of molecular simulation programs, including source code and demos.
- AmberTools. AmberTools consists of several independently developed packages that work well by themselves, and with Amber itself. It contains NAB (build molecules; run MD or distance geometry, using generalized Born, Poisson-Boltzmann or 3D-RISM implicit solvent models), antechamber and MCPB (create force fields for general organic molecules and metal centers), tleap and sleap (Basic preparation program for Amber simulations), sqm (semiempirical and DFTB quantum chemistry program), pbsa (Performs numerical solutions to Poisson-Boltzmann models), 3D-RISM (Solves integral equation models for solvation), ptraj and cpptraj (structure and dynamics analysis of trajectories), MMPBSA.py and amberlite (Energy-based analyses of MD trajectories)
- GROMACS. High performance toolkit for CPU/GPU-accelerated molecular dynamics simulation and setup. Can be used as a library or as an application with an intuitive Python interface. Developed, maintained and provided open-source by Standford University, United States.
- VFFDT. It consists in a user-friendly Visual Force Field Derivation Toolkit (VFFDT) to derive the force field parameters via simply clicking on the bond or angle in the 3D viewer, and we have further extended our previous program to support the Hessian matrix output from a variety of quantum mechanics (QM) packages, including Gaussian 03/09, ORCA 3.0, QChem, GAMESS-US, and MOPAC 2009/2012. In this toolkit, a universal VFFDT XYZ file format containing the raw Hessian matrix is available for all of the QM packages, and an instant force field parametrization protocol based on a semiempirical quantum mechanics (SQM) method is introduced. The new function that can automatically obtain the relevant parameters for zinc, copper, iron, etc., which can be exported in AMBER Frcmod format, has been added. Furthermore, our VFFDT program can read and write files in AMBER Prepc, AMBER Frcmod, and AMBER Mol2 format and can also be used to customize, view, copy, and paste the force field parameters in the context of the 3D viewer, which provides utilities complementary to ANTECHAMBER, MCPB, and MCPB.py in the AmberTools.Proposed by the School of Pharmaceutical Sciences, Wenzhou Medical University
- NAMD. (NAnoscale Molecular Dynamics). Free parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects.
- aMD. (accelerated Molecular Dynamics). enhanced-sampling method that improves the conformational space sampling by reducing energy barriers separating different states of a system. Distributed by the National Biomedical Computation Resource.
- MATCH. (Multipurpose Atom-Typer for CHARMM). Toolset of program libraries collectively titled multipurpose atom-typer for CHARMM (MATCH) for the automated assignment of atom types and force field paramters for molecular mechanics simulation of organic molecules. Developed by the Brooks lab, Michigan university.
- Desmond. Package to perform high-speed molecular dynamics simulations of biological systems on conventional computer clusters. Developed at D. E. Shaw Research.
- LAMMPS. Program for molecular dynamics. LAMMPS has potentials for soft materials (biomolecules, polymers), solid-state materials (metals, semiconductors) and coarse-grained or mesoscopic systems.
- MOLARIS-XG. MOLARIS-XG incorporates the methodologies of the former packages ENZYMIX and POLARIS. ENZYMIX is a macromolecular simulation program designed to study the functions of proteins ranging from ligand binding to free energy profiles of enzymatic reactions using the Empirical Valence Bond (EVB) approach and the Free Energy Perturbation (FEP) method. POLARIS is a fast converging computational software based on the Protein Dipoles-Langevin Dipoles (PDLD) approach, it is used for the calculation of free energies and electrostactic properties of molecules and macromolecules in solution through the evaluation of the permanent dipoles, induce dipoles, charges, dispersion contributions and hydrophobicity. Provided by the Warshel's group at the university of Southern California.
- OPENMD. Open source molecular dynamics to simulate liquids, proteins, nanoparticles, interfaces, and other complex systems using atom types with orientational degrees of freedom (e.g. sticky atoms, point dipoles, and coarse-grained assemblies).
- ORAC. Free open source program for Moleuclar Dynamics simulations. Maintained by the Florence university, Italy.
- AMMP VE. (Another Molecular Mechanics Program). Full-featured molecular mechanics, dynamics and modelling program that can manipulate both small molecules and macromolecules including proteins, nucleic acids and other polymers. Uses the VEGA ZZ interface. For MS Windows and Linux. Provided by the Drug Design Laboratory of the University of Milano.
- ACEMD (Accelerating bio-molecular simulations). Production bio-molecular dynamics (MD) software running on graphics processing units (GPUs) on NVIDIA graphics cards. ACEMD reads CHARMM/NAMD and AMBER input files. Distributed by Acellera. Free for 1 year for academic.
- CNS (Crystallography & NMR System). Provides a flexible multi-level hierachical approach for the most commonly used algorithms in macromolecular structure determination.
- Adun molecular simulation. Free biomolecular simulator developed at the Computational Biophysics and Biochemistry Laboratory, a part of the Research Unit on Biomedical Informatics of the UPF. It is distributed under the GNU General Public License.
- Tinker. Free, complete and general package for molecular mechanics and dynamics, with some special features for biopolymers.
- Force Field Explorer. Graphical user interface to the TINKER suite of molecular modeling tools.
- CHARMm. Commercial version of CHARMM with multiple graphical front ends.
- MacroModel. Commercial program for molecular modeling. Distributed by Schrodinger.
- MOIL. Public Domain Molecular Modeling Software, including energy calculations, energy minimization, molecular dynamics. Comes with a visualization program (zmoil) for graphic display of individual structures, dynamics, reaction paths and overlay of multiple structures, read PDB CRD DCD and (MOIL specific) PTH formatted files. For Windows, Mac and Linux.
- APBS. Adaptive Poisson-Boltzmann Solver (APBS) is a software for evaluating the electrostatic properties of nanoscale biomolecular systems.
- iAPBS. C/C++/Fortran interface to APBS. This interface enables access to most of APBS capability from within any C/C++ or Fortran code. In addition to the reference implementation, iAPBS/CHARMM, iAPBS/NAMD and iAPBS/Amber modules are also available. These modules extend CHARMM, NAMD and Amber functionality with APBS routines for electrostatic calculations. Provided by the McCammon Group, UCSD.
- Chemsol. Program to calculate solvation energies by using Langevin Dipoles (LD) model of the solvent and ab initio calculations. Also exists as a web service. Provided by the Warshel's group at the university of Southern California.
- BiKi. The BiKi Life Sciences suite involves several tools (e.g. accelerated binding/unbinding methods) for performing and analyzing MD specifically dedicated to medicinal chemists with the aim of simplifying drug discovery. Provided by BiKi Technologies, Italy.
- Abalone. General purpose molecular modeling program focused on molecular dynamics of biopolymers and molecular graphics. In addition, it can interact with external quantum chemical programs (NWChem, CP2K and PC GAMESS/Firefly. Provided by Agile Molecule.
- Ascalaph. General purpose molecular modeling suite that performs quantum mechanics calculations for initial molecular model development, molecular mechanics and dynamics simulations in the gas or in condensed phase. It can interact with external molecular modeling packages (MDynaMix, NWChem, CP2K and PC GAMESS/Firefly). Provided by Agile Molecule.
- HyperChem. Provides computational methods including molecular mechanics, molecular dynamics, and semi-empirical and ab-initio molecular orbital methods, as well as density functional theory.
- iHyperChem. Limited version of Professional HyperChem. This Level 1 version of iHyperChem allows creating and manipulating molecular systems and explore their structure. It also allows WiFi access to any Professional HyperChem server so that molecules, computations, and results can be transferred between the mobile client (iPhone or iPad) and the server. Provided by HyperChem.
- iHyperChem Free Version. Free Version of iHyperChem for iPad. Provided by HyperChem.
- Spartan. Provides computational methods including molecular mechanics, quantum mechanics, properties calculations (LogP, ovality, etc...), quantification of structural alignment using structure, chemical funtion descriptors or pharmacophore model, etc... Developed by Wavefunction, Inc.
- iSpartan. iSpartan is a versatile app for molecular modeling on the iPad, iPhone, and iPod Touch. Molecules are created by two-dimensional sketching and converted into a three-dimensional structure. Low-energy conformations can then be calculated and their geometries be queried. A database of 5,000 molecules (a subset of the Spartan Spectra and Properties Database, SSPD) can furthermore be accessed to obtain NMR and IR spectra, molecular orbitals, electrostatic potential maps, and other atomic and molecular properties. The database can be searched for substructures. Developed by Wavefunction, Inc.
- SCIGRESS. Desktop/server molecular modeling software suite that can apply a wide range of computational models to all types of molecular systems, from small organic molecules, to whole proteins, including linear scaling semiempirical quantum methods for protein optimization and ligand docking. Developed and distributed by Fujitsu, Ltd.
- TopoTools. TopoTools is a VMD plugin for manipulating topology information. It is meant to be a complementary tool to psfgen, which is very much optimized for building topologies for biomolecules. It makes access to the topology related data stored in VMD easily. It also has a number of high-level tools that allow reading and writing of topology file formats that cannot be parsed by the molfile plugins, parsing of parameter and residue database files for generation of complete input files for MD codes like LAMMPS and HOOMD-blue, and replicating or combining multiple systems. Developed by the Temple University, Philadelphia, USA.
- YASARA Dynamics. Adds support for molecular simulations to YASARA View/Model, Using the NOVA, YAMBER or AMBER force fields like AMBER. Provided by YASARA.
- Build model. Tool for creating protein models and their preparation for docking. Refine raw protein structure, add missing sidechains, assign protonation states of side chains at given pH, add missing hydrogen atoms, reconstruct crystallographically-related protein subunits and extract a reference ligand from the structure. Distributed by Moltech. For Windows and linux.
- Pdbfil. Automatically processes the protein coordinate data obtained from PDB for molecular calculations. Adds missing atoms, deletes unnecessary hetero-residues and water molecules, adds and optimizes hydrogen atoms. Atomic attributions like atomic charge or molecular force-field type are also automatically. Distributed by IMMD.
- Protein Preparation Wizard. Tool for correcting common structural problems and creating reliable, all-atom protein models. Distributed by Schrodinger.
- BALL. Biochemical Algorithms Library. Application framework in C++ designed for rapid software prototyping in the field of Computational Molecular Biology and Molecular Modeling. It provides an extensive set of data structures as well as classes for Molecular Mechanics, advanced solvation methods, comparison and analysis of protein structures, file import/export, and visualization. Free and opensource.
- pDynamo. pDynamo is an open source program library that has been designed for the simulation of molecular systems using quantum chemical (QC), molecular mechanical (MM) and hybrid QC/MM potential energy functions. Developed by the Institut de Biologie Structurale, Grenoble, France.
- Pcetk. Pcetk (a pDynamo-based continuum electrostatic toolkit) is a Python module extending the pDynamo library with a Poisson-Boltzmann continuum electrostatic model that allows for protonation state calculations in proteins. The module links pDynamo to the external solver of the Poisson-Boltzmann equation, extended-MEAD, which is used for the calculation of electrostatic energy terms. The calculation of protonation states and titration curves is done by using the module's own analytic or Monte Carlo routines or through an interface to the external sampling program, GMCT. Developed by the Institut de Biologie Structurale, Grenoble, France.
- MMTSB. Multiscale Modeling Tools for Structural Biology. Provides a collection of perl scripts for Structure preparation, Structure analysis, All-Atom Modeling, SICHO Lattice Modeling, Replica Exchange Sampling, Ensemble Computing and Structure Prediction.
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- pymacs. Python module for dealing with structure files and trajectory data from the GROMACS molecular dynamics package. It has interfaces to some gromacs functions and uses gromacs routines for command line parsing, reading and writing of structure files (pdb,gro,...) and for reading trajectory data (only xtc at the moment).
- PyRosetta. Interactive Python-based interface to the Rosetta molecular modeling suite. It enables users to design their own custom molecular modeling algorithms using Rosetta sampling methods and energy functions.
Web Services
- SwissParam. Provides topology and parameters for small organic molecules compatible with the CHARMM all atoms force field, for use with CHARMM and GROMACS.
- CHARMM-GUI. Provides (optimal, reasonable) CHARMM input files in a GUI fashion so that people can run the input on their machine, Helps people read and modify the input with their purposes, and provides also educational materials such as MM/MD lectures, as well as molecular animations. Provided by the University of Kansas.
- CHARMMing.org. CHARMMing contains an integrated set of tools for uploading structures, performing simulations, and viewing the results.
- LigParGen. Web-based service that provides OPLS-AA force field parameters for organic molecules or ligands. Beside PGR files, other ouput format includes parameters and topologies to be used with CHARMM, Gromacs, LAMMPS, CNS/X-PLOR, Q, DESMOND, BOSS, OpenMM and MCPRO. Molecules can be input in SMILES, MOL or PDB format with a maximum of 200 atoms. Provided also as a standalone program by the Department of Chemistry, University of Yale, United-States.
- LipidBuilder. A web-server based on a VMD plug-in and CHARMM force field to create, store and share lipid libraries. LipidBuilder automatically generates the topology and template of a given lipid. The lipid topology is created by combining the selected head group, extracted from a built-in library of structures and the provided hydrocarbon chains. Four different classes of hydrocarbons have been parametrized in the CHARMM force field: saturated, unsaturated, branched and cyclopropane. Developed by the EPFL, Lausanne, Switzerland.
- ParamChem. Provides topology and parameters for small organic molecules from CGenFF, for use with CHARMM and GROMACS. Provided by the University of Kentucky.
- PrimaDORAC. A free Web Interface for the Assignment of partial charges, chemical topology, and bonded parameters in small molecules to be used in molecular mechanics or molecular dynamics calculations. Provided by the University of Florence, Italy.
- MATCH server. The MATCH web server allows one to submit a molecule to generate both the topology and parameter file for a given small molecule. Molecule file formats: pdb, sdf, mol, mol2 are all accepted. Provided by the Brooks laboratory, University of Michigan.
- ProteinPrepare. a web application designed to interactively support the preparation of protein structures. Users can upload a PDB file, choose the solvent pH value, and inspect the resulting protonated residues and hydrogen-bonding network within a 3D web interface. Provided by Acellera, Ltd.
- MemBuilder. Prepares the initial configuration of a membrane model composed of different phospholipid molecules. This server is also dedicated to determine the lipid composition of each monolayer to support the asymmetry of the membrane bilayer. Provided by the Tarbiat Modares University.
- CABS-flex. Server for fast simulation of protein structure fluctuations. CABS-flex is a procedure for the simulation of structure flexibility of folded globular proteins. Using an input protein structure the CABS-flex outputs a set of protein models (reflecting the flexibility of the input structure, in all-atom PDB format) ready to use in structure-based studies of protein functions and interactions. Developed by the university of Warsaw.
- Dundee PRODRG2 Server. Converts coordinates for small molecules in PDB format to the following topology formats: GROMOS, GROMACS, WHAT IF, REFMAC5, CNS, O, SHELX, HEX and MOL2.
- PDB2PQR Server. Web server to convert PDB files into PQR files and perform an APBS calculation. Protein pKa are calculated using PROPKA.
- PROPKA. Web server to calculate pKa of protein titratable functions.
- PropKa. Web server to calculate pKa of protein titratable functions using PROPKA. Provided by the Drug Design Laboratory of the University of Milano.
- H++. Web server to compute pK values of ionizable groups in macromolecules and adds missing hydrogen atoms according to the specified pH of the environment.
- PDB_Hydro. Provides tools for mutating (change side-chains of a PDB file automatically, repair missing side-chains in a PDB file, construct polar hydrogen atoms and assig partial charges for electrostatic calculations) and solvating PDB files.
- POLYVIEW-MM. Web-based platform for animation and analysis of molecular simulations. Enables animation of trajectories generated by molecular dynamics and related simulation techniques, as well as visualization of alternative conformers, e.g. obtained as a result of protein structure prediction methods or small molecule docking.
- CLICK. Web server for superimposing the 3D structures of any pair of biomolecules (proteins, DNA, RNA, etc.). The server makes use of the Cartesian coordinates of the molecules with the option of using other structural features such as secondary structure, solvent accessible surface area and residue depth to guide the alignment. Help establishing protein relationships by detecting similarities in structural subdomains, domains and topological variants or to recognize conformational changes that may have occurred in structural domains or subdomains in one structure with respect to the other.
- SLITHER. Web server for generating contiguous conformations of substrate molecules entering into deep active sites of proteins or migrating across membrane transporters. Predicts whether a substrate molecule can crawl through an inner channel or a half-channel of proteins across surmountable energy barriers.
- AlloDeco. Server that implements a novel model for allostery. It computes the thermodynamic coupling between functional sites in proteins and then determines the contribution of specific interactions to that coupling. First, a 3-dimensional protein structure (PDB file) is transformed into a Gaussian Network Model. The coupled motions of two sites are then decomposed using Canonical Correlation Analysis. Finally, the statistical mechanics-based thermodynamic coupling function formalism is applied to identify interactions that mediate the thermodynamic coupling between the canonical pairs of motions. Developed at Weill Cornell Medical College, New York, United States.
- R.E.D. Server. Web service designed to automatically derive RESP and ESP charges, and to build force field libraries for new molecules/molecular fragments.
- Chemsol web service. Web service for the calculations of solvation free energies using the Langevin Dipoles (LD) solvation model, in which the solvent is approximated by polarizable dipoles fixed on a cubic grid.Also exists as a standalone program. Provided by the Warshel's group at the university of Southern California.
- MovieMaker. Web server that allows short (~10 sec), downloadable movies to be generated of protein dynamics. It accepts PDB files or PDB accession numbers as input and automatically outputs colorful animations covering a wide range of protein motions and other dynamic processes (simple rotation, morphing between two end conformers, short-scale, picosecond vibrations, ligand docking, protein oligomerization, mid-scale nanosecond (ensemble) motions and protein folding/unfolding). Note: MovieMaker is not a molecular dynamics server and does not perform MD calculations. Provided by the University of Alberta, Canada.
- Superpose. Protein superposition server, using a modified quaternion approach. From a superposition of two or more structures, it generates sequence alignments, structure alignments, PDB coordinates, RMSD statistics, Difference Distance Plots, and interactive images of the superimposed structures. Provided by the University of Alberta, Canada.
- ArbAlign. Web implmentation of the Kuhn-Munkres algorithm to optimally align two arbitrarily ordered isomers. Code and web interface provided by the Bucknell University, Lewisburg, USA.
Homology Modeling
Modeller, I-TASSER, LOMETS, SWISS-MODEL, SWISS-MODELRepository, Robetta, ...
Software
- Modeller. Software for producing homology models of protein tertiary structures, using a technique inspired by nuclear magnetic resonance known as satisfaction of spatial restraints. Maintained by Andrej Sali at the University of California, San Francisco. Free for academic use. Graphical user interfaces and commercial versions are distributed by Accelrys.
- I-TASSER. Internet service for protein structure and function predictions. Models are built based on multiple-threading alignments by LOMETS and iterative TASSER simulations. I-TASSER (as 'Zhang-Server') was ranked as the No 1 server in recent CASP7 and CASP8 experiments. Exists as a standalone package. Provided by the University of Michigan.
- LOMETS. (Local Meta-Threading-Server). On-line web service for protein structure prediction. It generates 3D models by collecting high-scoring target-to-template alignments from 8 locally-installed threading programs (FUGUE, HHsearch, MUSTER, PPA, PROSPECT2, SAM-T02, SPARKS, SP3). Exists as a standalone package. Provided by the University of Michigan.
- MPACK. (Modeling Package). Integrated protein modeling suite that currently handles comparative and ab initio modeling procedures. The objective of this suite is to systematically bring different steps (or programs) under one roof in order to facilitate rapid model generation with minimal user effort and to create a biological data-flow pipeline for large scale-scale modeling of protein sequences from genomic projects. Provided by the University of Texas.
- ProModel. Allows homology modeling from either a selected template or a user defined template. Modeling in manual mode allows mutation, excision, deletion, insertion of residues or insertion of a loop by selecting the start and end anchors. Automated homology modeling can be performed by reading in the template file obtained from a local BLAST. ProModel enables analysis of the target protein structure, active site and channels. Provided by VLife.
- SCRWL. Program for prediction of protein sice chains prediction, based on the Dunbrack backbone-dependent rotamer library. Provided by the Dunbrack Lab.
- Biskit. Free and open source modular, object-oriented Python library for structural bioinformatics research that wraps external programs (BLAST, T-Coffee and Modeller) into an automated workflow. Developed by the institut Pasteur.
- ModPipe. Completely automated software pipeline that can calculate protein structure models for a large number of sequences with almost no manual intervention. In the simplest case, it takes as input a sequence identifier and a configuration file and produces one or more comparative models for that sequence. Free and open source software. Maintained by Andrej Sali at the University of California, San Francisco.
- RaptorX. Protein structure prediction program developed by Xu group, with a particular focus on the alignment of distantly-related proteins with sparse sequence profile and that of a single target to multiple templates. Currently, RaptorX consists of four major modules: single-template threading, alignment quality assessment, multiple-template threading and fragment-free approach to free modeling. Also exists as a web service.
- Prime. Fully-integrated protein structure prediction program, providing graphical interface, sequence alignment, secondary structure prediction, homology modeling, protein refinement, loop-prediction, and side-chain prediction. Developed by Schrödinger.
- ProSide. Predicts protein sidechain conformation. Since the residue-substitution by the target amino-acid sequence is possible, ProSide can be used also for simple homology modeling, in case there are neither insertion nor deletion. Can perform global optimization calculation of a complex, by putting ligand to a binding site, and optimizing positions and conformations of ligand and amino-acid sidechains. Distributed by IMMD.
- CABS. Versatile reduced representation tool for molecular modeling, including: de novo folding of small proteins, comparative modeling (especially in cases of poor templates) and structure prediction based on sparse experimental data. Developed by the Warsaw University.
Web services and databases
- SWISS-MODEL. Fully automated protein structure homology-modeling server, accessible via the ExPASy web server, or from the program DeepView (Swiss Pdb-Viewer).
- SWISS-MODEL Repository. Database of annotated three-dimensional comparative protein structure models generated by the fully automated homology-modelling pipeline SWISS-MODEL.
- Robetta. Web server. Rosetta homology modeling and ab initio fragment assembly with Ginzu domain prediction.
- ModWeb. Server for Protein Structure Modeling based on the Modeller program. Maintained by Andrej Sali at the University of California, San Francisco.
- I-TASSER. Internet service for protein structure and function predictions. Models are built based on multiple-threading alignments by LOMETS and iterative TASSER simulations. I-TASSER (as 'Zhang-Server') was ranked as the No 1 server in recent CASP7 and CASP8 experiments. Exists as a standalone package. Provided by the University of Michigan.
- RaptorX web server. Protein structure prediction web server developed by Xu group, with a particular focus on the alignment of distantly-related proteins with sparse sequence profile and that of a single target to multiple templates. Currently, RaptorX consists of four major modules: single-template threading, alignment quality assessment, multiple-template threading and fragment-free approach to free modeling. Due to limited computational power, this server offers the first three modules for regular usage. Also exists as a standalone program.
- TIP database. The Target Informatics Platform (TIP) database contains more than 195,000 high resolution protein structures and homology models, with annotated small molecule binding sites, covering major drug target families including proteases, kinases, phosphatases, phosphodiesterases, nuclear receptors, and GPCRs. The TIP database automatically and self-consistently updates itself, possibly including proprietary sequence and structure data. Developed and maintained by Eidogen-Sertanty, Inc.
- iProtein. iPad application providing access to the Eidogen-Sertanty's Target Informatics Platform (TIP).
- ModBase. Database of three-dimensional protein models calculated by comparative modeling. The models are derived by ModPipe, an automated modeling pipeline relying on the programs PSI-BLAST and MODELLER. The database also includes fold assignments and alignments on which the models were based. MODBASE also contains information about putative ligand binding sites, SNP annotation, and protein-protein interactions.
- ModEval. Model evaluation server for protein structure models. Maintained by Andrej Sali at the University of California, San Francisco.
- ModLoop. Web server for automated modeling of loops in protein structures. The server relies on the loop modeling routine in MODELLER that predicts the loop conformations by satisfaction of spatial restraints, without relying on a database of known protein structures. Maintained by Andrej Sali at the University of California, San Francisco.
- Protinfo ABCM. The Protinfo web server consists of a series of discrete modules that make predictions of, and provide information about, protein folding, structure, function, interaction, evolution, and design by applying computational methodologies developed by the Samudrala Computational Biology Research Group.
- PMP. (Protein Model Portal). Gives access to various models computed by comparative modeling methods provided by different partner sites, and provides access to various interactive services for model building, and quality assessment. Provided by the Swiss Institute of BioInformatics and the University of Basel.
- HHpred. Web server for homology detection & structure prediction by HMM-HMM comparison.
- CPHmodels. Protein homology modeling server. The template recognition is based on profile-profile alignment guided by secondary structure and exposure predictions. Maintained by the Center for Biological Sequence Analysis, Denmmark.
- GeneSilico Metaserver. Gateway to various methods for protein structure prediction, including primary structure, seconday structure, transmembrane helices, disordered regions, disulfide bonds, nucleic acid binding residues in proteins and tertiary structure. Maintained by the Bujnicki laboratory in IIMCB, Warsaw, Poland.
- QUARK. Internet service for ab initio protein folding and protein structure prediction, which aims to construct the correct protein 3D model from amino acid sequence only. QUARK models are built from small fragments (1-20 residues long) by replica-exchange Monte Carlo simulation under the guide of an atomic-level knowledge-based force field. QUARK was ranked as the No 1 server in Free-modeling (FM) in CASP9. Since no global template information is used in QUARK simulation, the server is suitable for proteins which are considered without homologous templates. Provided by the University of Michigan.
- SuperLooper. SuperLooper provides an online interface for the automatic, quick and interactive search and placement of loops in proteins. Loop candidates are selected from a database (LIMP) comprising ~ 180.000 loops of membrane proteins or, alternatively, from (LIP) containing ~ 513.000.00 segments of water-soluble proteins with lengths up to 35 residues. In addition to several filtering criteria regarding structural and sequence features, the software allows for placing the loop within the predicted membrane-water interface. Provided by Charité Berlin, Structural Bioinformatics Group.
- PEP-FOLD. De novo approach aimed at predicting peptide structures from amino acid sequences. This method, based on structural alphabet SA letters to describe the conformations of four consecutive residues, couples the predicted series of SA letters to a greedy algorithm and a coarse-grained force field. Developed by the University of Paris Diderot.
- FoldX. A whole package for protein modeling and design. Originally focused on the impact of mutation on protein structure stability based on an original force field strongly linked to mutagenesis data. It was developed by at the European Molecular Biology Laboratory in Heidelberg and at Center for Genomic Regulation in Barcelona. The FoldX Suite is available through academic or commercial license.
- LOMETS. (Local Meta-Threading-Server). On-line web service for protein structure prediction. It generates 3D models by collecting high-scoring target-to-template alignments from 8 locally-installed threading programs (FUGUE, HHsearch, MUSTER, PPA, PROSPECT2, SAM-T02, SPARKS, SP3). Exists as a standalone package. Provided by the University of Michigan.
- ESyPred3D. Automated homology modeling web server in which lignments are obtained by combining, weighting and screening the results of several multiple alignment programs. The final three dimensional structure is built using the modeling package MODELLER.
- MolProbity. Web service for all-atom structure validation for macromolecular crystallography. Maintained by the Richardson Lab, Duke University.
- PSiFR. (Protein Structure and Function predicton Resource) provides integrated tools for protein tertiary structure prediction and structure and sequence-based function annotation. The web portal provides access to TASSER, TASSER-Lite and MetaTASSER and DBD-Hunter, and the enzyme function inference engine EFICAz2.
- 3D-Jigsaw. Automated system to build three-dimensional models for proteins based on homologues of known structure.
- Geno3D. Automatic modeling of proteins three-dimensional structure using comparative protein structure modelling by spatial restraints (distances and dihedral) satisfaction. Provided by the Pole Bioinformatique Lyonnais.
- VADAR. (Volume, Area, Dihedral Angle Reporter) is a compilation of more than 15 different algorithms and programs for analyzing and assessing peptide and protein structures from their PDB coordinate data to quantitatively and qualitatively assess protein structures determined by X-ray crystallography, NMR spectroscopy, 3D-threading or homology modelling. Provided by the University of Alberta, Canada.
- phyre. (Protein Homology/analogY Recognition Engine). Automated 3D model building using profile-profile matching and secondary structure. Provided by the Structural Bioinformatics group, Imperial College London.
- HMMSTR/Rosetta. Predicts the structure of proteins from the sequence : secondary, local, supersecondary, and tertiary. Provided by the Depts of Biology & Computer Science, Rensselaer Polytechnic Institute
- GPCRautomodel. Web service that automates the homology modeling of mammalian olfactory receptors (ORs) based on the six three-dimensional (3D) structures of G protein-coupled receptors (GPCRs) available so far and (ii) performs the docking of odorants on these models, using the concept of colony energy to score the complexes. Provided by INRA.
- FALC-Loop. Web server for protein loop modeling using a fragment assembly and analytical loop closure method.
- IntFOLD. Web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction.
- HOMODELLER. Web server to predict protein 3D structure (PDB coordinates) from its primary sequence file by homology modelling. Provided by the University of Alberta, Canada.
- PEPstr. Web server to predict the tertiary structure of small peptides with sequence length varying between 7 to 25 residues. The prediction strategy is based on the realization that β-turn is an important and consistent feature of small peptides in addition to regular structures. Provided by the Bioinformatics Centre, Institute of Microbial Technology, Chandigarh.
Binding site prediction
MED-SuMo, TRAPP, CAVER, sc-PDB, CASTp, Pocketome, 3DLigandSite, metaPocket, PockDrug, ...
Software
- MED-SuMo. Program for macromolecules surface similarity detection. Searches into 3D databases, find similar binding surfaces and generate 3D superpositions based on common surface chemical features and similar shape. Can be used for site mining, drug repurposing and site classification at PDB scale. Distributed by MEDIT.
- TRAPP. TRAnsient Pockets in Proteins (TRAPP) is a web server for the analysis of transient binding pockets in proteins. Contrarily to many tools, it is not intended for ligand binding pocket identification per se, but rather to predict significant changes in the spatial and physicochemical properties of a given pocket that may arise due to the protein's flexibility (both backbone and side chains). Several capabilities of visualization and analysis have been developed and are provided by the Molecular and Cellular Modeling group at Heidelberg Institute for Theoretical Studies, Germany.
- CAVER. Software tool for analysis and visualisation of tunnels and channels in protein structures. Provided by the Masaryk University.
- fpocket. Open source protein pocket (cavity) detection algorithm based on Voronoi tessellation. Developed in the C programming language and currently available as command line driven program. fpocket includes two other programs (dpocket & tpocket) that allow you to extract pocket descriptors and test own scoring functions respectively. Also contains a druggability prediction score.
- GHECOM. Program for finding multi-scale pockets on protein surfaces using mathematical morphology. Free open source.
- LIGSITEcsc. Program for the automatic identification of pockets on protein surface using the Connolly surface and the degree of conservation.
- SURFNET. Generates surfaces and void regions between surfaces from coordinate data supplied in a PDB file.
- SiteHound. Identifies ligand binding sites by computing interactions between a chemical probe and a protein structure. The input is a PDB file of a protein structure, the output is a list of “interaction energy clusters” corresponding to putative binding sites.
- ICM-PocketFinder. Binding site predictor based on calculating the drug-binding density field and contouring it at a certain level. Provided by Molsoft.
- SiteMap. Program for binding site identification. Distributed by Schrodinger.
- MSPocket. Orientation independent program for the detection and graphical analysis of protein surface pockets. A MSPocket plugin for PyMOL provides a graphical user interface for runing MSPocket and render its results in PyMOL. It is included in the download. Free and open source.
- POCASA. (POcket-CAvity Search Application). Automatic web service that implements the algorithm named Roll which can predict binding sites by detecting pockets and cavities of proteins of known 3D structure. Maintained by the Hokkaido University.
- Phosfinder. Method for the prediction of phosphate-binding sites in protein structures. provided by the University of Rome.
- VOIDOO. Software to find cavities and analyse volumes.
- FunFOLDQA. Program to assess the quality ligand binding site residue predictions based on 3D models of proteins. Free program written in java. Developped by the University of Reading.
- LISE. Free and open source program for ligand Binding Site Prediction Using Ligand Interacting and Binding Site-Enriched Protein Triangles. Exists as a web service. Provided by the Institute of Biomedical Sciences, Academia Sinica.
- PDBinder. Free program for the identification of small ligand-binding sites in a protein structure. webPDBinder searches a protein structure against a library of known binding sites and a collection of control non-binding pockets. Exists as a web service. Provided by the University of Roma 2, Italy.
- eFindSite. Ligand binding site prediction and virtual screening algorithm that detects common ligand binding sites in a set of evolutionarily related proteins identified by 10 threading/fold recognition methods. Exists as a web service. Provided by the Louisiana State University, Computational Systems Biology Group.
- POVME. Free and open source program for measuring binding-pocket volumes. Developed by the National Biomedical Computation Resource.
- SiteEngine. Program to predict regions that can potentially function as binding sites. The methods is based on recognition of geometrical and physico-chemical environments that are similar to known binding sites. Exists as a web service. Provided by the structural Bioinformatics group at Tel-Aviv University.
- SVILP_ligand. General method for discovering the features of binding pockets that confer specificity for particular ligands. Provided by the Computational Bioinformatics Laboratory, Imperial College London.
Databases
- sc-PDB. Annotated Database of Druggable Binding Sites from the Protein DataBank. Provided by the university of Strasbourg.
- CASTp. Computed Atlas of Surface Topography of proteins. Provides identification and measurements of surface accessible pockets as well as interior inaccessible cavities, for proteins and other molecules. castP server uses the weighted Delaunay triangulation and the alpha complex for shape measurements.
- Pocketome. Encyclopedia of conformational ensembles of all druggable binding sites that can be identified experimentally from co-crystal structures in the Protein Data Bank.
- PDBe motifs and Sites. Can be used to examine the characteristics of the binding sites of single proteins or classes of proteins such as Kinases and the conserved structural features of their immediate environments either within the same specie or across different species.
- LigASite. Dataset of biologically relevant binding sites in protein structures. It consists of proteins with one unbound structure and at least one structure of the protein-ligand complex. Both a redundant and a non-redundant (sequence identity lower than 25%) version is available.
- PROtein SURFace ExploreR. Contains information about structural similarities with respect to the query surfaces. A pocket search algorithm detected 48,347 potential ligand binding sites from the 9,708 non-redundant protein entries in the PDB database. All-against-all structural comparison was performed for the predicted sites, and the similar sites with the Z-score ≥ 2.5 were selected. These results can be accessed by the PDB code or ligand name.
- fPOP. Footprinting protein functional surfaces by comparative spatial patterns. Database of the protein functional surfaces identified by shape analysis.
- PDBSITE. Database on protein active sites and their spatial environment. Provided by GeneNetworks.
- LigBase. Database of ligand binding proteins aligned to structural templates. The structural templates are taken from the PDB, 3D models of the aligned sequences are provided ModBase, and pairwise sequence alignments are provided by CE. Multiple Structural Alignments are built on the fly within LigBase from a series of pairwise alignments. Ligand diagrams are generated with the program Ligplot. Maintained by Andrej Sali at the University of California, San Francisco.
Web services
- 3DLigandSite. Automated method for the prediction of ligand binding sites. Provided by the Imperial London College.
- metaPocket. Meta server to identify pockets on protein surface to predict ligand-binding sites.
- PockDrug. A methodology tehat predicts pocket druggability, efficient on both; estimated pockets guided by the ligand proximity (extracted by proximity to a ligand from a holo protein structure using several thresholds) and estimated pockets not guided by the ligand proximity (based on amino atoms that form the surface of potential binding cavities).. Developed and maintained by the University Paris-Diderot, France.
- PocketQuery. Protein-protein interaction (PPI) inhibitor starting points from PPI structure. Quickly identify a small set of residues at a protein interface that are suitable starting points for small-molecule design. Provided by the University of Pittsburgh.
- PASS. Program for tentative identification of drug interaction pockets from protein structure.
- DEPTH. Web server to compute depth and predict small-molecule binding cavities in proteins
- fpocket web server. Open source protein pocket (cavity) detection algorithm based on Voronoi tessellation. Developed in the C programming language and currently available as command line driven program. fpocket includes two other programs (dpocket & tpocket) that allow you to extract pocket descriptors and test own scoring functions respectively. Also contains a druggability prediction score.
- Nucleos. Nucleos is a webserver for the identification of nucleotide-binding sites based on the concept of nucleotide modularity. Nucleos identifies binding sites for nucleotide modules (namely the nucleobase, the carbohydrate and the phosphate) and then combines them in order to build the complete binding sites for different types of nucleotides (e.g. ADP or FAD). Provided by the University of Roma 2, Italy.
- wwwPDBinder. Web server for the identification of small ligand-binding sites in a protein structure. webPDBinder searches a protein structure against a library of known binding sites and a collection of control non-binding pockets. Exists as a standalone program. Provided by the University of Roma 2, Italy.
- IsoMIF. IsoMIF identifies binding site molecular interaction field similarities between proteins. The IsoMIF Finder Interface allows you to identify binding site molecular interaction field (MIF) similarities between a query structure and a database of pre-calculated MIFs or you own custom PDB entries. Developed by the University of Sherbrooke, Canada.
- LISE. Ligand Binding Site Prediction Using Ligand Interacting and Binding Site-Enriched Protein Triangles. Exists as a standalone program. Provided by the Institute of Biomedical Sciences, Academia Sinica.
- eFindSite. Web server for ligand binding site prediction and virtual screening algorithm that detects common ligand binding sites in a set of evolutionarily related proteins identified by 10 threading/fold recognition methods. Exists as standalone program. Provided by the Louisiana State University, Computational Systems Biology Group.
- Active Site Prediction. Web server for computing the cavities in a given protein. Provided by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- GHECOM web server. Web server for finding multi-scale pockets on protein surfaces using mathematical morphology.
- LIGSITEcsc web server. Web server for the automatic identification of pockets on protein surface using the Connolly surface and the degree of conservation.
- ProBis. Web server for detection of structurally similar binding sites. Maintained by the National Institute of Chemistry, Ljubljana, Slovenia.
- ProBiS-CHARMMing. Web server for detection of structurally similar binding sites, plus minimization of predicted protein-ligand complexes and their interaction energy calculation. Maintained by the National Institute of Health, USA.
- FunFOLD. Web server to predict likely ligand binding site residues for a submitted amino acid sequence.
- CAVER. Software tool for analysis and visualisation of tunnels and channels in protein structures. Provided by the Masaryk University.
- SuMo. Screens the Protein Data Bank (PDB) for finding ligand binding sites matching your protein structure or inversely, for finding protein structures matching a given site in your protein. Provided freely by the Pole Bioinformatique Lyonnais.
- IBIS. (Inferred Biomolecular Interactions Server). For a given protein sequence or structure query, IBIS reports physical interactions observed in experimentally-determined structures for this protein. IBIS also infers/predicts interacting partners and binding sites by homology, by inspecting the protein complexes formed by close homologs of a given query.
- PocketDepth. Depth based algortihm for identification of ligand binding sites.
- Screen2. Tool for identifying protein cavities and computing cavity attributes that can be applied for classification and analysis.
- SiteHound-web. Identifies ligand binding sites by computing interactions between a chemical probe and a protein structure. The input is a PDB file of a protein structure, the output is a list of “interaction energy clusters” corresponding to putative binding sites. Maintained by the Sanchez lab, at the Mount Sinai School of Medicine, NY, USA.
- SiteComp. Web server providing three major types of analysis based on molecular interaction fields: binding site comparison, binding site decomposition and multi-probe characterization. Maintained by the Sanchez lab, at the Mount Sinai School of Medicine, NY, USA.
- ConCavity. Ligand binding site prediction from protein sequence and structure.
- SplitPocket. Prediction of binding sites for unbound structures.
- PepSite 2. Web service for the prediction of peptide binding sites on protein surfaces. Developed and maintained by the Russel Lab, University of Heidelberg.
- MolAxis. Web server for the identification of channels in macromolecules.
- PDBSiteScan. Tool for search for functional sites in protein tertiary structures. Developed in collaboration with Institute of Cytology and Genetics, Novosibirsk.
- MultiBind. (Multiple Alignment of Protein Binding Sites). Prediction tool for protein binding sites. Users input a set of protein-small molecule complexes and MultiBind predicts the common physio-chemical patterns responsible for protein binding. Exists as a standalone program. Provided by the structural Bioinformatics group at Tel-Aviv University.
- SiteEngine. Web service to predict regions that can potentially function as binding sites. The methods is based on recognition of geometrical and physico-chemical environments that are similar to known binding sites. Exists as a standalone program. Provided by the structural Bioinformatics group at Tel-Aviv University.
Docking
Autodock, DOCK, GOLD, SwissDock, DockingServer, 1-ClickDocking, ...
Software
- Autodock. Free open source EA based docking software. Flexible ligand. Flexible protein side chains. Maintained by the Molecular Graphics Laboratory, The Scripps Research Institute, la Jolla.
- DOCK. Anchor-and-Grow based docking program. Free for academic usage. Flexible ligand. Flexible protein. Maintained by the Soichet group at the UCSF.
- GOLD. GA based docking program. Flexible ligand. Partial flexibility for protein. Product from a collaboration between the university of Sheffield, GlaxoSmithKline plc and CCDC.
- Glide. Exhaustive search based docking program. Exists in extra precision (XP), standard precision (SP) and virtual High Throughput Screening modes. Ligand and protein flexible. Provided by Schrödinger.
- Itzamna. Itzamna is a docking program, identifying active compounds for a given target. You can upload a protein and a docking is performed, either against an in-house database containing more than a million active compounds, or against a user-defined library. Provided by Mind The Byte.
- SCIGRESS. Desktop/server molecular modeling software suite employing linear scaling semiempirical quantum methods for protein optimization and ligand docking. Developed and distributed by Fujitsu, Ltd.
- GlamDock. Docking program based on a Monte-Carlo with minimization (basin hopping) search in a hybrid interaction matching / internal coordinate search space. Part of the Chil2 suite. Open for general research.
- FlexAID. A small-molecule docking algorithm that accounts for target side-chain flexibility and utilizes a soft scoring function. The pairwise energy parameters were derived from a large dataset of true positive poses and negative decoys from the PDBbind database through an iterative process using Monte Carlo simulations. Precompiled Linux, MacOS and Windows versions are made available by the University of Sherbrooke, Canada.
- GEMDOCK. Generic Evolutionary Method for molecular DOCKing. Program for computing a ligand conformation and orientation relative to the active site of target protein==== Docking - Software ====
- iGEMDOCK. Graphic environment for the docking, virtual screening, and post-screening analysis. Free for non commercial researches. For Windows and Linux.
- HomDock. Progam for similarity-based docking, based on a combination of the ligand based GMA molecular alignment tool and the docking tool GlamDock. Part of the Chil2 suite. Open for general research.
- ICM. Docking program based on pseudo-Brownian sampling and local minimization. Ligand and protein flexible. Provided by MolSoft.
- FlexX, Flex-Ensemble (FlexE). Incremental build based docking program. Flexible ligand. Protein flexibility through ensemble of protein structure. Provided by BioSolveIT.
- Fleksy. Program for flexible and induced fit docking using receptor ensemble (constructed using backbone-dependent rotamer library) to describe protein flexibility. Provided by the Centre for Molecular and Biomolecular Informatics, Radboud University Nijmegen.
- FITTED. (Flexibility Induced Through Targeted Evolutionary Description). Suite of programs to dock flexible ligands into flexible proteins. This software relies on a genetic algorithm to account for flexibility of the two molecules and location of water molecules, and on a novel application of a switching function to retain or displace water molecules and to form potential covalent bonds (covalent docking) with the protein side-chains. Part of the Molecular FORECASTER package and FITTED Suite. Free for an academic site license (excluding cluster).
- FORECASTER. Standalone interface that contains applications to perform docking and more. It includes the FITTED docking program, the sites of metabolism prediction IMPACTS, and the accessory programs to work with the proteins and the ligands. It comes with a java-based graphical interface that integrated all the program into workflows. Provided by Molecular Forecaster Inc.
- VLifeDock. Multiple approaches for protein - ligand docking. Provides three docking approches: Grid based docking, GA docking and VLife's own GRIP docking program. Several scoring functions can be used: PLP score, XCscore and Steric + Electrostatic score. Available for Linux and Windows. Provided by VLife.
- ParaDockS. (Parallel Docking Suite). Free, open source program, for docking small, drug-like molecules to a rigid receptor employing either the knowledge-based potential PMF04 or the empirical energy function p-Score.
- Molegro Virtual Docker. Protein-ligand docking program with support for displaceable waters, Induced-fit-docking, user-defined constraints, molecular alignment, ligands-based screening, and KNIME workflow integration. Distributed by Qiagen.
- DAIM-SEED-FFLD. Free open source fragment-based docking suite. The docking is realized in three steps. DAIM (Decomposition And Identification of Molecules) decomposes the molecules into molecular fragments that are docked using SEED (Program for docking libraries of fragments with solvation energy evaluation). Finally, the molecules are reconstructed ''in situ'' from the docked fragments using the FFLD program (Program for fragment-based flexible ligand docking). Developed and maintained by the Computational Structural Biology of ETH, Zurich, Switzerland.
- Autodock Vina. MC based docking software. Free for academic usage. Flexible ligand. Flexible protein side chains. Maintained by the Molecular Graphics Laboratory, The Scripps Research Institute, la Jolla.
- VinaMPI. Massively parallel Message Passing Interface (MPI) program based on the multithreaded virtual docking program AutodockVina. Free and open source. Provided by the University of Tennessee.
- FlipDock. GA based docking program using FlexTree data structures to represent a protein-ligand complex. Free for academic usage. Flexible ligand. Flexible protein. Developed by the Department of Molecular Biology at the Scripps Research Institute, la Jolla.
- PharmDock. A protein pharmacophore-based docking program. PharmDock and a PyMOL plugin are made freely available by the Purdue University, West Lafayette, USA.
- FRED. FRED performs a systematic, exhaustive, nonstochastic examination of all possible poses within the protein active site, filters for shape complementarity and pharmacophoric features before selecting and optimizing poses using the Chemgauss4 scoring function. Provided by OpenEye scientific software.
- POSIT. POSIT uses the information from bound ligands to improve pose prediction. Using a combination of approaches, including structure generation, shape alignment and flexible fitting, a ligand of interest is compared to bound ligands and its similarity to such both guides the nature of the applied algorithm and produces an estimate. Both 2D and 3D similarity measures are used in this reliability index. Provided by OpenEye scientific software.
- HYBRID. Docking program similar to FRED, except that it uses the Chemical Gaussian Overlay (CGO) ligand-based scoring function. Provided by OpenEye scientific software.
- idock. Free and open source multithreaded virtual screening tool for flexible ligand docking for computational drug discovery. Developed by the Chinese university of Hong Kong.
- POSIT. Ligand guided pose prediction. POSIT uses bound ligand information to improve pose prediction. Using a combination of several approaches, including structure generation, shape alignment and flexible fitting, it produces a predicted pose whose accuracy depends on similarity measures to known ligand poses. As such, it produces a reliability estimate for each predicted pose. In addition, if provided with a selection of receptors from a crystallographic series, POSIT will automatically determine which receptor is best suited for pose prediction. Provided by OpenEye scientific software.
- Rosetta Ligand. Monte Carlo minimization procedure in which the rigid body position and orientation of the small molecule and the protein side-chain conformations are optimized simultaneously. Free for academic and non-profit users.
- Surflex-Dock. Docking program based on an idealized active site ligand (a protomol), used as a target to generate putative poses of molecules or molecular fragments, which are scored using the Hammerhead scoring function. Distributed by Tripos.
- CDocker. CHARMm based docking program. Random ligand conformations are generated by molecular dynamics and the positions of the ligands are optimized in the binding site using rigid body rotations followed by simulated annealing. Provided by Accelrys.
- LigandFit. CHARMm based docking program. Ligand conformations generated using Monte-Carlo techniques are initially docked into an active site based on shape, followed by further CHARMm minimization. Provided by Accelrys.
- rDock. Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. Free and open source.Developed by the University of Barcelona.
- KIN. Kin is a blind-docking technology. All potential cavities of a given protein are predicted, and a query molecule is docked inside each of them, sorting results by scoring function. Distributed by Mind The Byte.
- Lead Finder. program for molecular docking, virtual screening and quantitative evaluation of ligand binding and biological activity.Distributed by Moltech. For Windows and linux.
- YASARA Structure. Adds support for small molecule docking to YASARA View/Model/Dynamics using Autodock and Fleksy. Provided by YASARA.
- ParaDockS. ParaDockS includes algorithms for protein-ligand docking and is organized that every newly developed scoring function can be immediately implemented. Furthermore, interaction-based classifier, trained on a target-specific knowledge base can be used in a post-docking filter step. An implementation and validation of target-biased scoring methods within the open-source docking framework is implemented. developed and provided free of charge by the University of Halle-Wittenberg, Germany.
- GalaxyDock. Protein-ligand docking program that allows flexibility of pre-selected side-chains of ligand. Developed by the Computational Biology Lab, Department of Chemistry, Seoul National University.
- MS-Dock. Free multiple conformation generator and rigid docking protocol for multi-step virtual ligand screening.
- BetaDock. Molecular docking simulation software based on the theory of Beta-complex.
- ADAM. Automated docking tool. Can be used for vHTS. Distributed by IMMD.
- hint!. (Hydropathic INTeractions). Estimates LogP for modeled molecules or data files, numerically and graphically evaluates binding of drugs or inhibitors into protein structures and scores DOCK orientations, constructs hydropathic (LOCK and KEY) complementarity maps that can be used to predict a substrate from a known receptor or protein structure or to propose the hydropathic structure from known agonists or antagonists, and evaluates/predicts effects of site-directed mutagenesis on protein structure and stability.
- DockVision. Docking package including Monte Carlo, Genetic Algorithm, and database screening docking algorithms.
- PLANTS. (Protein-Ligand ANT System). Docking algorithm based on a class of stochastic optimization algorithms called ant colony optimization (ACO). In the case of protein-ligand docking, an artificial ant colony is employed to find a minimum energy conformation of the ligand in the binding site. These ants are used to mimic the behavior of real ants and mark low energy ligand conformations with pheromone trails. The artificial pheromone trail information is modified in subsequent iterations to generate low energy conformations with a higher probability. Developed by the Konstanz university.
- EADock. Hybrid evolutionary docking algorithm with two fitness functions, in combination with a sophisticated management of the diversity. EADock is interfaced with the CHARMM package for energy calculations and coordinate handling.
- EUDOC. Program for identification of drug interaction sites in macromolecules and drug leads from chemical databases.
- FLOG. Rigid body docking program using databases of pregenerated conformations. Developed by the Merck Research Laboratories.
- Hammerhead. Automatic, fast fragment-based docking procedure for flexible ligands, with an empirically tuned scoring function and an automatic method for identifying and characterizing the binding site on a protein.
- ISE-Dock. Docking program which is based on the iterative stochastic elimination (ISE) algorithm.
- ASEDock. Docking program based on a shape similarity assessment between a concave portion (i.e., concavity) on a protein and the ligand. Developed by yoka Systems.
- HADDOCK. HADDOCK (High Ambiguity Driven biomolecular DOCKing) is an approach that makes use of biochemical and/or biophysical interaction data such as chemical shift perturbation data resulting from NMR titration experiments, mutagenesis data or bioinformatic predictions. First developed from protein-protein docking, it can also be applied to protein-ligand docking. Developed and maintained by the Bijvoet Center for Biomolecular Research, Netherlands.
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- Autodock Vina plugin for PyMOL. Allows defining binding sites and export to Autodock and VINA input files, doing receptor and ligand preparation automatically, starting docking runs with Autodock or VINA from within the plugin, viewing grid maps generated by autogrid in PyMOL, handling multiple ligands and set up virtual screenings, and set up docking runs with flexible sidechains.
- GriDock. Virtual screening front-end for AutoDock 4. GriDock was designed to perform the molecular dockings of a large number of ligands stored in a single database (SDF or Zip format) in the lowest possible time. It take the full advantage of all local and remote CPUs through the MPICH2 technology, balancing the computational load between processors/grid nodes. Provided by the Drug Design Laboratory of the University of Milano.
- DockoMatic. GUI application that is intended to ease and automate the creation and management of AutoDock jobs for high throughput screening of ligand/receptor interactions.
- BDT. Graphic front-end application which control the conditions of AutoGrid and AutoDock runs. Maintained by the Universitat Rovira i Virgili,
Web services
- SwissDock. SwissDock, a web service to predict the molecular interactions that may occur between a target protein and a small molecule.
- DockingServer. DockingServer offers a web-based, easy to use interface that handles all aspects of molecular docking from ligand and protein set-up.
- 1-Click Docking. Free online molecular docking solution. Solutions can be visualized online in 3D using the WebGL/Javascript based molecule viewer of GLmol. Provided by Mcule.
- Blaster. Public access service for structure-based ligand discovery. Uses DOCK as the docking program and various ZINC Database subsets as the database.Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Docking At UTMB. Web-driven interface for performing structure-based virtual screening with AutoDock Vina. Maintained by the Watowich lab at the University of Texas Medical Branch.
- Blind Docking Server. A web-based tool to run molecular docking on the whole surface of the protein. The calculation are based on a customized version of Autodock Vina. Results and analyses can be explored on-line or downloaded. Some services are free, other are cost-based. Developed by the Bioinformatics and High Performance Computing Research group at the Universidad Católica San Antonio de Murcia (UCAM), Spain.
- Pardock. All-atom energy based Monte Carlo, rigid protein ligand docking, implemented in a fully automated, parallel processing mode which predicts the binding mode of the ligand in receptor target site. Maintained by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- FlexPepDock. High-resolution peptide docking (refinement) protocol, implemented within the Rosetta framework. The input for this server is a PDB file of a complex between a protein receptor and an estimated conformation for a peptide.
- PatchDock. Web server for structure prediction of protein-protein and protein-small molecule complexes based on shape complementarity principles.
- MEDock. Maximum-Entropy based docking web server for efficient prediction of ligand binding sites.
- BSP-SLIM. Web service for blind molecular docking method on low-resolution protein structures. The method first identifies putative ligand binding sites by structurally matching the target to the template holo-structures. The ligand-protein docking conformation is then constructed by local shape and chemical feature complementarities between ligand and the negative image of binding pockets. Provided by the University of Michigan.
- BioDrugScreen. Computational drug design and discovery resource and server. The portal contains the DOPIN (Docked Proteome Interaction Network) database constituted by millions of pre-docked and pre-scored complexes from thousands of targets from the human proteome and thousands of drug-like small molecules from the NCI diversity set and other sources. The portal is also a server that can be used to (i) customize scoring functions and apply them to rank molecules and targets in DOPIN; (ii) dock against pre-processed targets of the PDB; and (iii) search for off-targets. Maintained by the laboratory of Samy Meroueh at the Center for Computational Biology and Bioinformatics at the Indiana University School of Medicine.
- GPCRautomodel. Web service that automates the homology modeling of mammalian olfactory receptors (ORs) based on the six three-dimensional (3D) structures of G protein-coupled receptors (GPCRs) available so far and (ii) performs the docking of odorants on these models, using the concept of colony energy to score the complexes. Provided by INRA.
- kinDOCK. Allows comparative docking of ligands into the ATP-binding site of a protein kinase (target). A sequence alignment of the target and a protein kinase profile is performed using HMMER. It uses protein-protein superposition (automatically restricted to the ligand binding pocket) of the target three-dimensional structure with those of known complexes of protein kinases/ligands.
- iScreen. Web service for docking and screening the small molecular database on traditional Chinese medicine (TCM) on user's protein. iScreen is also implemented with the de novo evolution function for the selected TCM compounds using the LEA3D genetic algorithm
- idTarget. Web server for identifying biomolecular targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Maintained by the National Taiwan University.
- MetaDock. Online docking solution and docking results analysis service. Docking is done with GNU/GPL-licensed AutoDock v.4 and Dock6 under academic license
- Score. Allows to calculate some different docking scores of ligand-receptor complex that can be submitted as a whole file containing both interaction partners or as two separated files. The calculation phase is provided by VEGA. Provided by the Drug Design Laboratory of the University of Milano.
- Pose & Rank. Web server for scoring protein-ligand complexes. Provided by the laboratory of Andrej Sali.
- PLATINUM. Calculates hydrophobic properties of molecules and their match or mismatch in receptor–ligand complexes. These properties may help to analyze results of molecular docking.
Screening
Pharmer, Catalyst, PharmaGist, SwissSimilarity, Blaster, AnchorQuery, ...
Software
- Pharmer. Free open source pharmacophore search technology that can search millions of chemical structures in seconds.
- Catalyst. Pharmacophore Modeling and Analysis; 3D database building and searching; Ligand conformer generation and analysis tools; Geometric, descriptor-based querying; Shape-based screening. Distributed by Accelrys as part of Discovery Studio.
- PharmaGist. Freely available web server for pharmacophore detection. The download version includes virtual screening capability.
- LiSiCA. LiSiCA (Ligand Similarity using Clique Algorithm) is a ligand-based virtual screening software that searches for 2D and 3D similarities between a reference compound and a database of target compounds which should be represented in a Mol2 format. The similarities are expressed using the Tanimoto coefficients and the target compounds are ranked accordingly. A PyMol plu-in is freely available, too. Developed by the University of Ljubljana, Slovenia.
- LigandScout. Fully integrated platform for virtual screening based on 3D chemical feature pharmacophore models. Developed by inte:ligand.
- CHAAC. Chaac is a ligand-based virtual screening tool. It compares your molecule with a database of ligands, and outputs a list of candidates with similar chemical profile to that of your query. Developed by Mind the Byte.
- IK. This virtual screening tool allows to compare in 3D molecules according to their behaviour with their environment. It generates a list of compounds similar to your query as output including also the non-structural analogues. Developed by Mind the Byte.
- KIZIN. Kizin supports compound selection. Given an input protein present in the ChEMBL database, and an internal or external library of drug candidates, it performs a 2D virtual screening, selecting molecules in the library likely to exhibit activity for that protein. Developed by Mind the Byte.
- ACPC. (AutoCorrelation of Partial Charges) Open source tool for ligand-based virtual screening using autocorrelation of partial charges. ACPC uses a rotation-translation invariant molecular descriptor.
- ChemCom. a computer application which facilitates searching and comparing chemical libraries. ChemCom aims to expedite the current, time consuming processes of comparing large, chemical databases. As such, this application can be used to speedup many processes including drug research and discovery. A free java web application is also available. Developed by the University of Kansas, USA.
- CoLibri. Assembles huge compound collections from multiple sources and various input formats into a virtual screening library, removes duplicates, assesses the distribution of physico-chemical properties of the compounds and makes selections/filter based on any property-threshold, molecules name-pattern or presence/absence of a particular substructure motif. Generates fragments library. Modifies molecules or fragments for generating, transforming and general handling of virtual screening libraries. Distributed by BioSolveIT.
- Corina. Generates 3D structures for small and medium sized, drug-like molecules. Distributed by Molecular Networks.
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- PL-PatchSurfer2. Virtual screening program using local surface matching between ligand and protein pocket. Zernike descriptor allows to calculate complementarity of the shape and physicochemical complementarity of both partners. Developed and provided as binary executable by Perdue University, United States.
- DecoyFinder. Graphical tool which helps finding sets of decoy molecules for a given group of active ligands. It does so by finding molecules which have a similar number of rotational bonds, hydrogen bond acceptors, hydrogen bond donors, logP value and molecular weight, but are chemically different, which is defined by a maximum Tanimoto value threshold between active ligand and decoy molecule MACCS fingerprints. Optionally, a maximum Tanimoto value threshold can be set between decoys in order to assure chemical diversity in the decoy set.
- DOVIS. (DOcking-based VIrtual Screening). Tool for virtual screening of chemical databases containing up to millions of small, drug-like compounds. The designed docking-based virtual screening pipeline uses the AutoDock 4.0 program as its docking engine and is integrated into an HPC environment. Its purpose is to remove many technical and administrative complexities involved in employing AutoDock for large scale virtual screening. Developed by the Biotechnology High Performance Computing Software Applications Institute.
- PyRX. Virtual Screening software for Computational Drug Discovery that can be used to screen libraries of compounds against potential drug targets. PyRx includes docking wizard with easy-to-use user interface which makes it a valuable tool for Computer-Aided Drug Design. PyRx also includes chemical spreadsheet-like functionality and visualization engine that are essential for Rational Drug Design. AutoDock 4 and AutoDock Vina are used as a docking software. Free and open source. For Windows, Linux and Mac OSX.
- MOLA. Free software for Virtual Screening using AutoDock4/Vina in a computer cluster using non-dedicated multi-platform computers. MOLA is integrated on a customized Live-CD GNU/LINUX operating system and is distributed as a MOLA.iso file. Distributed by BioChemCore.
- NNScore. Neural-Network-Based Scoring Function for the Characterization of Protein-Ligand Complexes. Reads PDBQT files as input. Developed by the University of California San Diego.
- WinDock. Program for structure-based drug discovery tasks under a uniform, user friendly graphical interface for Windows-based PCs. Combines existing small molecule searchable three-dimensional (3D) libraries, homology modeling tools, and ligand-protein docking programs in a semi-automatic, interactive manner, which guides the user through the use of each integrated software component. Developed by the Howard University College of Medecine.
- DockoMatic. GUI application that is intended to ease and automate the creation and management of AutoDock jobs for high throughput screening of ligand/receptor interactions.
- MolSign. Program for pharmacophore identification and modeling. Can be used for querying databases as a pharmacophore based search. Provided by VLife.
- Spectrophores. Converts three-dimensional molecular property data (electrostatic potentials, molecular shape, lipophilicity, hardness and softness potentials) into one-dimensional spectra independent of the position and orientation of the molecule. It can be used to search for similar molecules and screen databases of small molecules. Open source software developed by Silicos.
- Shape-it. free open source shape-based alignment tool by representing molecules as a set of atomic Gaussians. Open source software developed by Silicos.
- Align-it. (Formerly Pharao). Pharmacophore-based tool to align small molecules. The tool is based on the concept of modeling pharmacophoric features by Gaussian 3D volumes instead of the more common point or sphere representations. The smooth nature of these continuous functions has a beneficent effect on the optimisation problem introduced during alignment. Open source software developed by Silicos.
- Open3DALIGN. Command-line tool aimed at unsupervised molecular alignment. Alignments are computed in an atom-based fashion (by means of a novel algorithm inspired to the LAMDA algorithm by Richmond and co-workers), in a pharmacophore-based fashion using Pharao as the alignment engine, or finally using a combination of the latter two methods. Free open source software. For Windows, Linux and Mac.
- Molegro Virtual Docker. The built-in Docking Template tool makes it possible to perform ligand-based screening by flexibly aligning a number of ligands (and determine a score for their similarity) and to perform hybrid docking by guiding the docking simulation by combining the template similarity score with a receptor-based docking scoring function. Distributed by Qiagen.
- GMA (Graph based Molecular Alignment). Combined 2D/3D approach for the fast superposition of flexible chemical structures. Part of the Chil2 suite. Open for general research.
- Fuzzee. Allows the identification of functionally similar molecules, based upon functional and structural groups or fragments. Part of the Chil2 suite. Open for general research.
- REDUCE. (Formerly FILTER). Tool to filter compounds from libraries using descriptors and functional groups. Part of the Molecular FORECASTER package, from Molecular Forecaster Inc.
- SELECT. (Selection and Extraction of Libraries Employing Clustering Techniques). Creates subset of libraries by diversity or similarity using clustering techniques. Part of the Molecular FORECASTER package.
- AutoclickChem. Computer program capable of performing click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compounds for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Exists as a web server. Distributed by the National Biomedical Computation Resource.
- REACTOR. (Rapid Enumeration by Automated Combinatorial Tool and Organic Reactions). Creates library of molecules by combining fragment libraries from a defined reaction, or from a generic attachment point on the fragments. Part of the Molecular FORECASTER package.
- gateio. (Fingerprints for Ligands and Proteins). Provides a common reference framework for comparing molecules, using GRID Molecular Interaction Fields (MIFs). The fingerprints are characterised by quadruplets of pharmacophoric features and can be used for ligand-ligand, ligand-receptor, and receptor-receptor comparison. In addition, the quadruplets can be used to align molecules, and a more detailed comparison of the GRID MIF overlap calculated. When the template is a ligand, this enables ligand-based virtual screening and alignment. When the template is a receptor site, this enables structure-based screening and pose prediction. Provided by Molecular Discovery.
- GASP. Genetic Algorithm Similarity Program. Generates pharmacophores using a genetic algorithm. Distributed by Tripos.
- Tuplets. Pharmacophore-based virtual screening. Distributed by Tripos.
- KeyRecep. Estimates the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. Can be used to estimate activities and vHTS. Distributed by IMMD.
- LigPrep. 2D to 3D structure conversions, including tautomeric, stereochemical, and ionization variations, as well as energy minimization and flexible filters to generate ligand libraries that are optimized for further computational analyses. Distributed by Schrodinger.
- Balloon. Free command-line program that creates 3D atomic coordinates from molecular connectivity via distance geometry and confomer ensembles using a multi-objective genetic algorithm. The input can be SMILES, SDF or MOL2 format. Output is SDF or MOL2.
- Epik. Enumerates ligand protonation states and tautomers in biological conditions. Distributed by Schrodinger.
- Bluto. Performs energy minimization and energy analysis of protein or protein-ligand complexes by using force field, for structural optimization of docking models of multiple ligands onto a protein. Provides tabular reports of the energy analysis such as the interaction energy. Suitable for vHTS. Distributed by IMMD.
- VSDMIP. Virtual Screening Data Management on an Integrated Platform. Comes with a PyMOL graphical user interface. Developed by the Centro de Biología Molecular Severo Ochoa.
Web services
- SwissSimilarity. Web tool for rapid ligand-based virtual screening of small to unprecedented ultralarge libraries of small molecules. Screenable compounds include drugs, bioactive and commercial molecules, as well as 205 million of virtual compounds readily synthesizable from commercially available synthetic reagents. Predictions can be carried out on-the-fly using six different screening approaches, including 2D molecular fingerprints as well as superpositional and fast nonsuperpositional 3D similarity methodologies. SwissSimilarity is part of a large initiative of the SIB Swiss Institute of Bioinformatics to provide online tools for computer-aided drug design, such as SwissDock, SwissBioisostere or SwissTargetPrediction with which it can interoperate, and is linked to other well-established online tools and databases. User interface and backend have been designed for simplicity and ease of use, to provide proficient virtual screening capabilities to specialists and nonexperts in the field. The SwissSimilarity website, developed by the Molecular Modeling Group of SIB Swiss Institute of Bioinformatics, is accessible free of charge or login.
- Blaster. Public access service for structure-based ligand discovery. Uses DOCK as the docking program and various ZINC Database subsets as the database.Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- AnchorQuery. Specialized pharmacophore search for targeting protein-protein interactions. Interactively search more than 20 million readily synthesizable compounds all of which contain an analog of a specific amino acid. Provided by the University of Pittsburgh.
- istar. Free web platform for large-scale protein-ligand docking based on the idock software. The web site can be downloaded and installed independently from GitHub. Developed by the Chinese university of Hong Kong.
- istar. Free web platform for large-scale protein-ligand docking based on the idock software. This link corresponds to the web site code that can be installed independently. Developed by the Chinese university of Hong Kong.
- GFscore. GFscore is a ranked-based consensus scoring function based on the five scoring functions : FlexX Score, G_Score, D_Score, ChemScore, and PMF Score available in TRIPOS Cscore module. The aim is to eliminate as many molecules as possible from proprietary in house database after a Virtual Library Screening (VLS) using TRIPOS FlexX for docking and the TRIPOS Cscore module for scoring. Developped and maintained by the Institute for Structural Biology and Microbiology, Marseille, France.
- Aggregator Advisor. Free web service to suggest molecules that aggregate or may aggregate under biochemical assay conditions. The approach is based on the chemical similarity to known aggregators, and physical properties. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- e-LEA3D. Searches the FDA approved drugs either by keyword or by substructure. Also builds combinatorial library of molecules.
- Combinatorial library design. Web server providing a click chemistry engine to connect one or more reactants on a central core (scaffold).
- eDesign. Web server providing a de novo drug design engine to create new molecules either from scratch (lead-hopping) or based on a user-defined scaffold on which R-groups have to be optimized. Alternatively, the same tool can be used to screen a library of molecules. The sructure-based function is based on the program PLANTS. Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.
- GFscore. Web server to discriminate true negatives from false negatives in a dataset of diverse chemical compounds using a consensus scoring in a Non-Linear Neural Network manner. The global scoring function is a combination of the five scoring functions found in the Cscore package from Tripos Inc.
- ZincPharmer. Free online interactive pharmacophore search software for screening the ZINC database. ZINCPharmer can import LigandScout and MOE pharmacophore definitions as well as perform structure-based pharmacophore elucidation.
- PUMA. Free web services that help at visulizing chemical space by computing molecular properties of pharmaceutical relevance, such as Murcko scaffolds, and performing diversity analysis. Developed and provided by the Department of Pharmacy of Universidad Nacional Autónoma de Mexico, Mexico.
- SimDOCK. Allows rapid selection of ligands fitting the active site of the submitted protein by superposition of its three-dimensional structure with those of known complexes of protein/ligands of the family.
- pep:MMs:MIMIC. Web-oriented tool that, given a peptide three-dimensional structure, is able to automate a multiconformers three-dimensional similarity search among 17 million of conformers calculated from 3.9 million of commercially available chemicals collected in the MMsINC database.
- wwLig-CSRre. Online Tool to enrich a bank a small compound with compounds similar to a query.
- AURAmol. Web service taking a candidate 2D or 3D molecular shape and use it to search for similarly shaped molecules in large databases. Provided by the University of York.
- SiMMap. Web server statistically deriving site-moiety map with several anchors, based on the target structure and several docked compounds. Each anchor includes three elements: a binding pocket with conserved interacting residues, the moiety composition of query compounds and pocket-moiety interaction type (electrostatic, hydrogen bonding or van der Waals). Compound highly agreeing with anchors of site-moiety map are expected to activate or inhibit the target protein.
- ShaEP. Free program to superimpose two rigid 3D molecular structure models, based on shape and electrostatic potentials, and computes a similarity index for the overlay. It can be used for the virtual screening of libraries of chemical structures against a known active molecule, or as a preparative step for 3D QSAR methods.
- AutoclickChem. Web server to perform click-chemistry reactions in silico. AutoClickChem can be used to produce large combinatorial libraries of compounds for use in virtual screens. As the compounds of these libraries are constructed according to the reactions of click chemistry, they can be easily synthesized for subsequent testing in biochemical assays. Exists as a stand alone program. Maintained by the National Biomedical Computation Resource.
Target prediction
PatchSearch, IXCHEL, CABRAKAN, SwissTargetPrediction, SEA, CSNAP, ...
Software
- PatchSearch. PatchSearch implements local searching for similar binding sites on protein surfaces with a controlled amount of flexibility. It is based on product graphs to represent all possible matchings between two structures. Developed and provided as an R package by Univeristé Paris-Diderot, France.
- IXCHEL. Ixchel is a protein-based biological activity prediction application. The input molecule (in SDF or SMILE) is docked in a database of over 9 000 protein cavities. Distributed by Mind The Byte.
- CABRAKAN. Cabrakan is a 2D ligand-based virtual profiling application. It compares molecules through their 2D-fingerprints and predicts their biological activity. Distributed by Mind The Byte.
- HURAKAN. Hurakan is a 3D ligand-based virtual profiling application. It compares molecules according to their interaction with their environment, without superimposition, to obtain compounds with different structures but predicted with similar bioactivity. Distributed by Mind The Byte.
- MolScore-Antivirals. Expert system to identify and prioritise antiviral drug candidates. Developed by PharmaInformatic, Germany.
- MolScore-Antibiotics. Expert system to identify and prioritise antibacterial drug candidates. Developed by PharmaInformatic, Germany.
Web services
- SwissTargetPrediction. Online tool to predict the targets of bioactive small molecules in human and other vertebrates. This is useful to understand the molecular mechanisms underlying a given phenotype or bioactivity, to rationalize possible side-effects or to predict off-targets of known molecules. Provided by the Molecular Modeling group of the Swiss Institute of BioInformatics.
- SEA. SEA (Similarity ensemble approach) relates proteins based on the set-wise chemical similarity among their ligands. It can be used to rapidly search large compound databases, build cross-target similarity maps and predict possible targets of a small molecule. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- CSNAP. CSNAP (Chemical Similarity Network Analysis Pull-down) is a computational approach for compound target identification based on network similarity graphs. Query and reference compounds are populated on the network connectivity map and a graph-based neighbor counting method is applied to rank the consensus targets among the neighborhood of each query ligand. Developed in the Torres lab at the University of California, Los Angeles (UCLA).
- PPB. PPB (Polypharmacology Browser) searches through 4613 groups of at least 10 bioactive molecules with documented activity against a biological target, as listed in ChEMBL, to identify analogs of any query molecule using six different fingerprints and four fingerprint combination (HyperSpace), and displays results groups by targets as lists of bioactive compounds. Provided by the Dept. of Chemistry and Biochemistry of the University of Bern, Switzerland.
- ChemProt. The ChemProt 2.0 server is a ressource of annotated and predicted chemical-protein interactions. The server is a compilation of over 1 100 000 unique chemicals with biological activity for more than 15000 proteins. ChemProt can assist in the in silico evaluation of small molecules (drugs, environmental chemicals and natural products) with the integration of molecular, cellular and disease-associated proteins complexes. Provided by the Technical University of Denmark, and the University Paris Diderot.
- SuperPred. Webservice for drug classification and target prediction. The web-server translates a user-defined molecule into a structural fingerprint that is compared to about 6300 drugs, which are enriched by 7300 links to molecular targets of the drugs, derived through text mining followed by manual curation. Provided by the Institute of Molecular Biology and Bioinformatics, Charité - University Medicine Berlin.
- PASSonline. (Prediction of Activity Spectra for Substances). Web service for evaluating the general biological potential of an organic drug-like molecule, based on the comparison of the user's compound to a database of 260,000 of drug-like biologically active compounds using the Multilevel Neighborhoods of Atoms (MNA) structure descriptors. Provided by the Orekhovich Institute of Biomedical Chemistry
- Target Hunter of Small Molecule. Web portal for predicting the therapeutic potential of small organic molecules based on chemogenomic database. Created and maintained by Prof. Xiang-Qun (Sean) Xie’s laboratory
- HitPick. Web server that facilitates the analysis of chemical screenings by identifing hits and predicting their molecular targets. For target prediction, HitPick applies an approach that combines two 2D molecular similarity based methods: a simple 1-Nearest-Neighbour similarity searching and a machine learning method based on Laplacian-modified naive Bayesian models. provided by the Helmholtz Center Munich, germany.
- Molinspiration bioactivity score. Score a compound for its ability to be GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, enzyme inhibitor. Based on Bayesian statistics to compare structures of representative ligands active on the particular target with structures of inactive molecules and to identify substructure features (which in turn determine physicochemical properties) typical for active molecules. Provided by Molinspiration.
- ElectroShape Polypharmacology server. Web service to estimate polypharmacology profiles and side effects of compounds based on the molecular similarity concept. Developed and maintained by Alvaro Cortes Cabrera.
Ligand design
GANDI, LUDI, BREED, SwissBioisostere, VAMMPIRE, sc-PDB-Frag, e-LEA3D, eDesign, iScreen, ...
Software
- GANDI. Program for structure-based fragment-based ab initio (de novo) ligand design. Developed and distributed by the Computational Structural Biology group of prof. Amedeo Caflisch, Zurich University.
- LUDI. Program for automated structure-based drug design, using growing and linking approaches. Distributed by Accelrys as part of Discovery Studio.
- BREED. Program for ligand-based ligand design, by hybridization of known ligands. Distributed by Schrodinger.
- AutoT&T2. The Automatic Tailoring and Transplanting (AutoT&T) method is developed as a versatile computational tool for lead optimization as well as lead discovery in molecular-targeted drug design. This method detects suitable fragments on reference molecules, e.g. outputs of a virtual screening job in prior, and then transplants them onto the given lead compound to generate new ligand molecules. Then, binding affinities, synthetic feasibilities and drug-likeness properties are evaluated to select the promising candidates for further consideration. Standalone software and demo web version
- Allegrow. Program for structure-based fragment-based ligand design, based on growing and combinatorial approaches. Distributed by Boston De Novo Design.
- E-novo. Program for automated structure-based ligand design, using a combinatorial substitution of R-groups on the initial scaffold. Distributed by Accelrys as part of Discovery Studio.
- SILCS. The Site Identification by Ligand Competitive Saturation Method (SILCS) allows for location and estimation of affinities of chemical groups on a protein surface. It relies on MD of the macromolecular structure in an explicit solvent/small molecules environment, representative of chemical fragments of diverse properties (hydophobic, aromatic, with H-bonding capacity). Maps are so created indidcating favorable fragment-protein interactions, to be used for structure-based designed. Distributed by SilcsBio, LLC.
- BOMB. Program for structure-based fragment-based ligand design, based on a growing approach. Distributed by Cemcomco.
- ChIP. Program, based on a genetic algorithm, for the exploration of synthetically feasible small molecule chemical space from commercially available starting materials, directly toward medicinally relevancy, applying predictive computational QSAR models and physicochemical and structural filters. ChIP can take account of propriatary chemical reactions. Developed by Eidogen-Sertanty, Inc.
- ChemT. Open-source software for building chemical compound libraries, based on a specific chemical template. The compound libraries generated can then be evaluated, using several Virtual Screening tools like molecular docking or QSAR modelling tools. Distributed by BioChemCore.
- MEGA. Program for structure-based fragment-based ligand design, based on a EA approach. Distributed by Noesis Informatics (NSisToolkit).
- LigBuilder. Program for structure-based fragment-based ab initio ligand design, based on growing, linking and mutation approaches.
- LeadGrow. Provides features for focused combinatorial library generation and screening to grow a lead molecule and perform lead optimization. Provides Lipinski screen and activity prediction using pre-generated QSAR models. Provided by VLife.
- Phase. Program for ligand-based drug design using pharmacophore modeling. Distributed by Schrodinger.
- CombiGlide. Program for ligand-based drug design using ligand-receptor scoring, combinatorial docking algorithms, and core-hopping technology to design focused libraries and identify new scaffolds. Distributed by Schrodinger.
- CoLibri. Assembles huge compound collections from multiple sources and various input formats into a virtual screening library, removes duplicates, assesses the distribution of physico-chemical properties of the compounds and makes selections/filter based on any property-threshold, molecules name-pattern or presence/absence of a particular substructure motif. Generates fragments library. Modifies molecules or fragments for generating, transforming and general handling of virtual screening libraries. Distributed by BioSolveIT.
- ReCore. Replaces a given pre-defined central unit of a molecule (the core), by searching fragments in a 3D database for the best possible replacement, while keeping the rest of the query compound. Additionally, user-defined "pharmacophore" constraints can be employed to restrict solutions. Distributed by BioSolveIT.
- LigMerge. Program combining structures of known binders to generate similar but structurally distinct compounds that can be tested for binding. Free and open source. Distributed by the National Biomedical Computation Resource.
- LEGEND. Program for automated structure-based drug design, using an atom-based growing approach. Provided by IMMD.
- Autogrow. Ligand design using fragment-based growing, docking, and evolutionary techniques. AutoGrow uses AutoDock as the selection operator. Provided by the McCammon Group, UCSD.
- CrystalDock. Computer algorithm that aids the computational identification of molecular fragments predicted to bind a receptor pocket of interest. CrystalDock identifies the microenvironments of an active site of interest and then performs a geometric comparison to identify similar microenvironments present in ligand-bound PDB structures. Germane fragments from the crystallographic or NMR ligands are subsequently placed within the novel active site. These positioned fragments can then be linked together to produce ligands that are likely to bind the pocket of interest; alternatively, they can be joined to an inhibitor with a known or suspected binding pose to potentially improve binding affinity. Free and opensource. For Mac OSX, Linux and Windows XP. Developed by the National Biomedical Computation Resource.
- MED-Ligand. Computational fragment-based drug design protocol. Annotated fragments of PDB ligands (MED-Portions) are positioned with MED-SuMo in 3D in a binding site and hybridised with MED-Ligand. Leads are discovered and optimised by hybridisation of MED-Portion chemical moities. Distributed by MEDIT.
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- RACHEL. (Real-time Automated Combinatorial Heuristic Enhancement of Lead compounds). Structure-based all-purpose ligand refinement software package designed to combinatorially derivatize a lead compound to improve ligand-receptor binding. Developed by Drug Design Methodologies and distributed by Tripos.
- MCSS. CHARMm-based method for docking and minimizing small ligand fragments within a protein binding site. Distributed by Accelrys.
- DLD. Automated method for the creation of novel ligands, linking up small functional groups that have been placed in energetically favorable positions in the binding site of a target molecule (See MCSS).
- LoFT. Tool for focused combinatorial library design using a (ligand-based) weighted multiobjective scoring function based on physicochemical descriptors.
- ACD/Structure Design Suite. Helps chemists optimize the physicochemical properties of their compounds. The software suggests alternative substituents (at a site/sites on the molecule) to drive the property of choice in the desired direction. Helps adjust aqueous solubility, lipophilicity (logP or logD), or change the ionization profile (pKa) of molecules. Distributed by ACD/Labs.
- HSITE. Program for automated structure-based drug design, using fitting and clipping of planar skeleton.
- PRO_LIGAND. Program for automated structure-based drug design, using growing and linking approaches.
- BUILDER. Program for structure-based ab initio ligand design. Finds molecule templates that bind tightly to 'hot spots' in the target receptor, and then generate bridges to join these templates.
- CONCERTS. Program for structure-based ab initio ligand design. Fragments are move independently about a target active site during a molecular dynamics simulation and are linked together when the geometry between proximal fragments is appropriate.
- ADAPT. Program for structure-based ab initio ligand design based pn the DOCK docking software.
- CoG. Program for ligand-based ab initio ligand design, using a graph-based genetic algorithm.
- Flux. Program for ligand-based ligand design using a EA approach.
- LCT. Program for structure-based ligand design using a linking approach.
- Biogenerator. Program for structure-based design of macrolides using a biomimetic synthesis of substitutide macrolides approach.
- ilib diverse. Program for creating virtual libraries of drug-like organic molecules suitable for rational lead structure discovery. Ligands are designed by combining user-defined fragments according to state-of-the-art chemical knowledge. Generated compounds can be filtered according to a variety of physico-chemical filters. Developed by inte:ligand.
- EMIL. (Example Mediated Innovation for Lead evolution). Suggests chemical modifications to hits to turn them into bona fide leads. EMIL searches through its Knowledge Base looking for similar chemistry and how it was optimized for potency and bioavailability (Iientification of bioisosteric/bioanalogous structures, indication of empirical information of the modification, such as change in physicochemical, in vitro and in vivo effects, etc...). Distributed by CompuDrug.
- Legio. Indigo-based GUI application that exposes the combinatorial chemistry capabilities of Indigo. Free and open source. Distributed by GGA software.
Databases
- SwissBioisostere. Freely available database containing information on millions of molecular replacements and their performance in biochemical assays. It is meant to provide researchers in drug discovery projects with ideas for bioisosteric modifications of their current lead molecule, and to give access to the details on particular molecular replacements. Users can provide a molecular fragment and get possible replacements, along with the biological assays in which they were observed. Users can also provide a given molecular replacement and get the corresponding information. The data were created through detection of matched molecular pairs and mining bioactivity data in the ChEMBL database. Developed and maintained by Merck Serono and the Swiss Institute of BioInformatics.
- VAMMPIRE. (Virtually Aligned Matched Molecular Pairs Including Receptor Environment) matched molecular pairs database for structure-based drug design and optimisation. By building MMPs between PDBbind and ChEMBLdb ligands VAMMPIRE extrapolates the two-dimensional ChEMBLdb ligands to the assumed, three-dimensional binding mode and introduce the received binding information into the database. Provided by the Institute of Pharmaceutical Chemistry / Goethe University Frankfurt, Germany.
- sc-PDB-Frag. Database of protein-bound fragments to help selecting truely bioisosteric scaffolds. The database allows to (i) search fragment among PDB ligands or sketch it; (ii) define similarity rules to retrieve potential bioisosteres; (iii) score bioisosteres according to interaction pattern similarity; (iv) align bioisosteres to the reference scaffold; (v) Visualize the proposed alignment.
- Glide Fragment Library. Set of 441 unique small fragments (1-7 ionization/tautomer variants; 6-37 atoms; MW range 32-226) derived from molecules in the medicinal chemistry literature. The set includes a total of 667 fragments with accessible low energy ionization and tautomeric states and metal and state penalties for each compound from Epik. These can be used for fragment docking, core hopping, lead optimization, de novo design, etc. Provided by Schrödinger.
- FragmentStore. Fragment Store is a database, primarily designed for pharmacists, biochemists, and medical scientists but also researchers working in cognate disciplines like the fragment based drug design. It provides access to information about fragments of compounds with their properties (e.g. charge, hydrophobicity, binding site preferences). It allow the user to do statistical analysis of the fragments properties and binding site preferences. Moreover, the database supports to build an adequate fragment library for fragment based drug design. Provided by the Structural Bioinformatics Group of Charité Berlin.
Web services
- e-LEA3D. Invents ideas of ligand (scaffold-hopping) by the de novo drug design program LEA3D.
- eDesign. Web server providing a de novo drug design engine to create new molecules either from scratch (lead-hopping) or based on a user-defined scaffold on which R-groups have to be optimized. Alternatively, the same tool can be used to screen a library of molecules. The structure-based function is based on the program PLANTS. Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.
- iScreen. Web service for docking and screening the small molecular database on traditional Chinese medicine (TCM) on user's protein. iScreen is also implemented with the de novo evolution function for the selected TCM compounds using the LEA3D genetic algorithm
- 1-Click Scaffold Hop. Draw a reference structure to get new scaffolds using a ligand-based approach. Allows visualizing the similarity between the query and the identified scaffold. Provided by mcule.
- 3DLigandSite. Automated method for the prediction of ligand binding sites. Provided by the Imperial London College.
- PASS. Program for tentative identification of drug interaction pockets from protein structure.
- DEPTH. Web server to compute depth and predict small-molecule binding cavities in proteins
- VAMMPIRE-LORD. LORD (Lead Optimization by Rational Design) is a prediction tool based on the VAMMPIRE database (of matched molecular pairs) and using a atom-pair descriptor to represent the substitution environment. It operates on the principle that molecular transformations cause similar effects in similar substitution environments and is therefore able to extrapolate the knowledge of a given substitution effect to any similar system. LORD was implemented as an easy-to-use web server that guides the user step-by-step through the optimization process of a defined lead compound.
Binding free energy estimation
Hyde, X-score, NNScore, DSXONLINE, BAPPLserver, BAPPL-Zserver, CLiBE, ...
Software
- Hyde. Entirely new approach to assess binding affinities and contributions to binding of a complex, with a visual feedback at atomic detail. Hyde shows which regions of a complex contribute favorably and infavorably to the binding. Allows modifying a molecule interactively to optimize a complex and trigger new lead optimization ideas. Hyde is entirely based on physics-principles and has not been trained or calibrated on experimental data. Distributed by BioSolveIT.
- X-score. Program for computing the binding affinities of the given ligand molecules to their target protein. X-Score is released to the public for free.
- NNScore. Python script for computing binding free energies from PDBQT files of the receptor and the ligand, using a neural network approach. Free and open source. Developed by the McCammon Lab, UCSD.
Web services
- DSXONLINE. (Formerly DrugScoreONLINE). Web-based user interface for the knowledge-based scoring functions DSX.
- BAPPL server. Binding Affinity Prediction of Protein-Ligand (BAPPL) server computes the binding free energy of a non-metallo protein-ligand complex using an all atom energy based empirical scoring function.
- BAPPL-Z server. Binding Affinity Prediction of Protein-Ligand complex containing Zinc [ BAPPL-Z ] server computes the binding free energy of a zinc containing metalloprotein-ligand complex using an all atom energy based empirical scoring function.
- PreDDICTA. Predict DNA-Drug Interaction strength by Computing ΔTm and Affinity of binding.
- PharmaGist. Freely available web server for pharmacophore detection. The download version includes virtual screening capability.
- IC50-to-Ki converter. Computes Ki values from experimentally determined IC50 values for inhibitors of enzymes that obey classic Michaelis-Menten kinetics and of protein-ligand interactions
Databases
- CLiBE. Database containing information about Computed Ligand-Receptor Interaction Energy and other attributes such as energy components; ligand classification, functions and properties. Ligand structure is also included. Provided by the BioInformatics and Drug Design group of the National University of Singapore.
QSAR
cQSAR, SeeSAR, clogP, MOLEdb, ChemDB/Datasets, DatasetsfromtheMilanoChemometricsandQSARResearchGroup, OCHEM, E-Dragon, PatternMatchCounter, ...
Software
- cQSAR. A regression program that has dual databases of over 21,000 QSAR models. Distributed by BioByte.
- SeeSAR. Program for interactive, visual compound promotion and optimization. It include PD and PK parameters and can be linked to other modules for physicochemical and ADME. Distributed by Bio
- clogP. Program for calculating log Poct/water from structure. Distributed by BioByte.
- ClogP/CMR. Estimates Molar Refractivity and logP. Distributed by Tripos.
- Topomer CoMFA. 3D QSAR tool that automates the creation of models for predicting the biological activity or properties of compounds. Distributed by Tripos.
- QSARPro. QSAR software for evaluation of several molecular descriptors along with facility to build the QSAR equation (linear or non-linear regression) and use it for predicting the activities of test/new set of molecules. Performs 2D and 3D QSAR, and provides GQSAR, a group based QSAR approach establishing a correlation of chemical group variation at different molecular sites of interest with the biological activity. Works on LInux and Windows. Provided by VLife.
- MedChem Studio. (Formerly ClassPharmer). Cheminformatics platform supporting lead identification and prioritization, de novo design, scaffold hopping and lead optimization. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- Surflex-Sim. Performs the alignment of molecules by maximizing their three-dimensional similarity. Surflex-Sim uses a surface-based morphological similarity function while minimizing the overall molecular volume of the aligned structures. Distributed by Tripos.
- QSAR with CoMFA. Builds statistical and graphical models that relate the properties of molecules (including biological activity) to their structures. Several structural descriptors can be calculated, including EVA and the molecular fields of CoMSIA. Distributed by Tripos.
- Almond. 3D-QSAR approach using GRid-INdependent Descriptors (GRIND). Starting with a set of 3D structures, Almond employs GRID3 force field to generate Molecular Interaction Fields (MIFs). The information in the MIFs is transformed to generate information-rich descriptors independent of the location of the molecules within the grid. Distributed by Tripos.
- GALAHAD. GA-based program to develop pharmacophore hypotheses and structural alignments from a set of molecules that bind at a common site. No prior knowledge of pharmacophore elements, constraints, or molecular alignment is required. Distributed by Tripos.
- Molegro Data Modeller. A program for building regression or classification models, performing feature selection and cross-validation, principal component analysis, high-dimensional visualization, clustering, and outlier detection. Provided by Qiagen.
- Hologram QSAR (HQSAR). Program using molecular holograms and PLS to generate fragment-based structure-activity relationships. Unlike other 3D-QSAR methods, HQSAR does not require alignment of molecules.
- cQSAR. A regression program that has dual databases of over 21,000 QSAR models. Distributed by BioByte.
- McQSAR. Free program to generates quantitative structure-activity relationships (QSAR equations) using the genetic function approximation paradigm. For Windows and Linux.
- CheS-Mapper. CheS-Mapper (Chemical Space Mapper) is a 3D-viewer for chemical datasets with small compounds. The tool can be used to analyze the relationship between the structure of chemical compounds, their physico-chemical properties, and biological or toxic effects. CheS-Mapper embedds a dataset into 3D space, such that compounds that have similar feature values are close to each other. It can compute a range of descriptors and supports clustering and 3D alignment. It is an open-source Java application, based on the Java libraries Jmol, CDK, WEKA, and utilizes OpenBabel and R. Developed and proposed by the Universität Mainz, Germany.
- Open3DQSAR. Program aimed at high-throughput generation and chemometric analysis of molecular interaction fields (MIFs). Free open source software. For Windows, Linux and Mac.
- PaDEL-Descriptor. Free software to calculate molecular (1875) descriptors and (12) fingerprints. Can be used from command lines or GUI. Developed by the National University of Singapore.
- Codessa. Derives descriptors using quantum mechanical results from AMPAC. These descriptors are then used to develop QSAR/QSPR models.
- CDK Descriptor Calculator GUI. Free and open source GUI to CDK to calculate molecular descriptors.
- BlueDesc. Free and open source molecular descriptor calculator. Converts an MDL SD file into ARFF and LIBSVM format for machine learning and data mining purposes using CDK and JOELib2. Provided by the Tuebingen University.
- KeyRecep. Estimates the characteristics of the binding site of the target protein by superposing multiple active compounds in 3D space so that the physicochemical properties of the compounds match maximally with each other. Can be used to estimate activities and vHTS. Distributed by IMMD.
- OpenMolGRID. Uses a Grid approach to deal with large-scale molecular design and engineering problems. The methodology used relies on Quantitative Structure Property/Activity Relationships (QSPR/QSAR).
- Molconn-Z. Standard program for generation of Molecular Connectivity, Shape, and Information Indices for Quantitative Structure Activity Relationship (QSAR) Analyses.
- CODESSA Pro. Program for developing quantitative structure-activity/property relationships (QSAR/QSPR. Distributed by CompuDrug.
- MCASE. Machine learning approach to automatically evaluate compounds/activity data set and identify the structural features responsible for activity (biophores). It then creates organized dictionaries of these biophores and develops ad hoc local QSAR correlations. Distributed by MultiCASE.
- hint!. (Hydropathic INTeractions). Estimates LogP for modeled molecules or data files, numerically and graphically evaluates binding of drugs or inhibitors into protein structures and scores DOCK orientations, constructs hydropathic (LOCK and KEY) complementarity maps that can be used to predict a substrate from a known receptor or protein structure or to propose the hydropathic structure from known agonists or antagonists, and evaluates/predicts effects of site-directed mutagenesis on protein structure and stability.
- smirep. System for predicting the structural activity of chemical compounds.
Databases
- MOLE db. Molecular Descriptors Data Base is a free on-line database comprised of 1124 molecular descriptors calculated for hundreds of thousands of molecules.
- ChemDB/Datasets. Experimentally annotated subsets of the ChemDB for machine learning and searching experiments.
- Datasets from the Milano Chemometrics and QSAR Research Group. References Data Sets
- OCHEM Database. Online database of experimental measurements integrated with a modeling environment. User can submit experimental data or use the data uploaded by other users to build predictive QSAR models for physical-chemical or biological properties. Provided by eADMET GmbH and the Institute of Bioinformatics & Systems Biology at Helmholtz Zentrum München.
- ChemSAR. Free web-based pipelining platform for classification models of small molecules. It includes validation and standardization of chemical structures, calculation of descriptots (1D, 2D and FP), feature selection, model building and interpretation. Developed by the School of Pharmaceutical Sciences, Central South University, China.
- Chembench. Free portal that enables researchers to mine available chemical and biological data. It includes robust model builders, property and activity predictors, virtual libraries of available chemicals with predicted biological and drug-like properties, and special tools for chemical library design. Provided with registration by the Carolina Exploratory Center for Cheminformatics Research, USA.
Web services
- OCHEM. (Online Chemical Modeling Environment project). Online database of experimental measurements integrated with a modeling environment. User can submit experimental data or use the data uploaded by other users to build predictive QSAR models for physical-chemical or biological properties. Provided by eADMET GmbH and the Institute of Bioinformatics & Systems Biology at Helmholtz Zentrum München.
- E-Dragon. Online version of DRAGON, which is an application for the calculation of molecular descriptors developed by the Milano Chemometrics and QSAR Research Group. These descriptors can be used to evaluate molecular structure-activity or structure-property relationships, as well as for similarity analysis and highthroughput screening of molecule databases. Provided by the Virtual Computational Chemistry Laboratory.
- Pattern Match Counter. Counts Functional Groups (sub-structures) in molecules.
- Pattern Count Screen. Screens by Functional Groups.
- Partial Least Squares Regression (PLSR). Generates model construction and prediction of activity/property using the Partial Least Squares (PLS) regression technique. Provided by the Virtual Computational Chemistry Laboratory.
- XScore-LogP. Calculates the octanol/water partition coefficient for a drug, based on a feature of the X-Score program.
- 3-D QSAR. 3-D QSAR MODELS DATABASE for Virtual Screening. users can process their own molecules by drawing or uploading them to the server and selecting the target for the virtual screening and biological activity prediction.
- MOLFEAT. Web service to compute molecular fingerprints and molecular descriptors of molecules from their 3D structures, and for computing activity of compounds of specific chemical types against selected targets based on published Quantitative Structure-Activity Relationship (QSAR) models. Currently covers 1,114 fingerprints, 3,977 molecular descriptors, and 23 QSAR models for 16 chemical types against 14 targets. Maintained by the University of Singapore.
ADME Toxicity
QikProp, VolSurf, GastroPlus, ALOGPS, OSIRISPropertyExplorer, SwissADME, PACT-F, TOXNET, LeadscopeToxicityDatabase, ...
Software
- QikProp. Provides rapid ADME predictions of drug candidates. Distributed by Schrodinger.
- VolSurf. Calculate ADME Properties and Create Predictive ADME Models. Distributed by Tripos.
- GastroPlus. Simulates the oral absorption, pharmacokinetics, and pharmacodynamics for drugs in human and preclinical species. The underlying model is the Advanced Compartmental Absorption and Transit (ACAT) model. Distributed plu Simulation Plus, Inc.
- MedChem Studio. Cheminformatics platform for computational and medicinal chemists supporting lead identification and optimization, in silico ligand based design, and clustering/classifying of compound libraries. It is integrated with MedChem Designer and ADMET Predictor. Distributed by Simulation Plus, Inc.
- ADMET Predictor. Software for advanced predictive modeling of ADMET properties. ADMET Predictor estimates a number of ADMET properties from molecular structures, and is also capable of building predictive models of new properties from user's data via its integrated ADMET Modeler module. Distributed by Simulations Plus, Inc.
- DDDPlus. Models and simulates the in vitro dissolution of active pharmaceutical ingredients (API) and formulation excipients dosed as powders, tablets, capsules, and swellable or non-swellable polymer matrices under various experimental conditions. Distributed by Simulations Plus, Inc.
- ADMEWORKS ModelBuilder. Builds QSAR/QSPR models that can later be used for predicting various chemical and biological properties of compounds. Models are based on values of physicochemical, topological, geometrical, and electronic properties derived from the molecular structure, and can be imported into ADMEWORKS Predictor.
- ADMEWORKS Predictor. QSAR based Virtual (in silico) screening system intended for simultaneous evaluation of the properties of compounds.
- MedChem Designer. Tool that combines molecule drawing features with a few free ADMET property predictions from ADMET Predictor. Distributed by Simulations Plus, Inc.
- IMPACT-F. Expert system to estimate oral bioavailability of drug-candidates in humans. IMPACT-F is composed of several QSAR models to predict oral bioavailability in humans. Developed by PharmaInformatic, Germany.
- MolScore-Drugs. Expert system to identify and prioritise drug candidates. Developed by PharmaInformatic, Germany.
- Natural product likeness calculator. Calculates Natural Product(NP)-likeness of a molecule, i.e. the similarity of the molecule to the structure space covered by known natural products. NP-likeness is a useful criterion to screen compound libraries and to design new lead compounds. Free and open source.
- ADMET Modeler. Integrated module of ADMET Predictor that automates the process of making high quality predictive structure-property models from sets of experimental data. It works seamlessly with ADMET Predictor structural descriptors as its inputs, and appends the selected final model back to ADMET Predictor as an additional predicted property. Distributed by Simulations Plus, Inc.
- Metabolizer. Enumerates all the possible metabolites of a given substrate, predicts the major metabolites and estimates metabolic stability. It can be used for the identification of metabolites by MS mass values, discovery of metabolically sensitive functionalities and toxicity prediction, and provide information related to the environmental effects of chemicals by bacterial degradation. Provided by ChemAxon.
- ACD/PhysChem Suite. Predicts basic physicochemical properties, like pKa, logP, logD, aqueous solubility and other molecular properties in seconds, usr a fragment-based models. Distributed by ACD/Labs.
- ACD/ADME Suite. Predicts of ADME properties from chemical structure, like Predict P-gp specificity, oral bioavailability, passive absorption, blood brain barrier permeation, distribution, P450 inhibitors, substrates and inhibitors, maximum recommended daily dose, Abraham-type (Absolv) solvation parameters. Distributed by ACD/Labs.
- ACD/Tox Suite. Collection of software modules that predict probabilities for basic toxicity endpoints. Several modules including hERG Inhibition, CYP3A4 Inhibition, Genotoxicity, Acute Toxicity, Aquatic Toxicity, Eye/Skin Irritation, Endocrine System Disruption, and Health Effects. Distributed by ACD/Labs.
- ACD/DMSO Solubility. Predicts solubility in DMSO solution. Distributed by ACD/Labs.
- Filter-it. Command-line program for filtering molecules with unwanted properties out of a set of molecules. The program comes with a number of pre-programmed molecular properties that can be used for filtering. Open source software distributed by Silicos.
- Virtual LogP. Bernard Testa's Virtual logP calculator. Provided by the Drug Design Laboratory of the University of Milano.
- FAF-Drugs2. Free package for in silico ADMET filtering. Distributed by the university of Paris Diderot.
- Discovery Studio TOPKAT Software. Cross-validated models for the assessments of chemical toxicity from chemical's molecular structure. Distributed by Accelrys.
- Discovery Studio ADMET Software. The ADMET Collection provides components that calculate predicted absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties for collections of molecules. Distributed by Accelrys.
- PreADME. Calculates molecular descriptors. Predicts Drug-likeness. ADME predictions.
- Molcode Toolbox. Molcode Toolbox allows prediction of medicinal and toxicological endpoints for a large variety of chemical structures, using proprietary QSAR models.
- KOWWIN - EPI Suite. Estimates the log octanol-water partition coefficient of chemicals using an atom/fragment contribution method. Distributed by the EPA~s Office of Pollution Prevention Toxics and Syracuse Research Corporation (SRC) as part of the EPI Suite. For Windows.
- ADRIANA.Code. Program to calculate physico-chemical properties of small molecules: number of H-bonds donor and acceptors, logP, logS, TPSA, dipole moment, polarizability, etc. Distributed by Molecular Networks.
- Derek Nexus. Predicts toxicity properties using QSAR and other expert knowledge rules. Distributed by Lhasa Limited.
- Meteor. Predicts metabolic fate of chemicals using other expert knowledge rules in metabloism. Distributed by Lhasa Limited.
- OncoLogic. Evaluates the likelihood that a chemical may cause cancer, using SAR analysis, experts decision mimicking and knowledge of how chemicals cause cancer in animals and humans. Distributed for free by the US Environmental Protection Agency (EPA).
- HazardExpert Pro. Predicts the toxicity of organic compounds based on toxic fragments. Distributed by CompuDrug.
- MetabolExpert. Predicts the most common metabolic pathways in animals, plants or through photodegradation. Distributed by CompuDrug.
- MEXAlert. Identifies compounds that have a high probability of being eliminated from the body in a first pass through the liver and kidney. Distributed by CompuDrug.
- PrologP/PrologD. Predicts the logP/logD values using a combination of linear and neural network methods. Distributed by CompuDrug.
- pKalc. Program for predicting acidic and basic pKa. Distributed by CompuDrug.
- Leadscope. Estimates toxiticy using QSAR. Distributed by Leadscope.
- COMPACT. Identifies potential carcinogenicity or toxicities mediated by CYP450s.
- CASETOX. Uses MCASE to predict toxicity. Distributed by MultiCASE.
- META. Predicts metabolic paths of molecules. Distributed by MultiCASE.
- PK-Sim. Predicts ADMET properties. Distributed by Bayer technology Services.
- SimCYP. The SimCYP Population-based ADME Simulator is a platform for the prediction of drug-drug interactions and pharmacokinetic outcomes in clinical populations. Distributed by SimCYP.
- SimCYP for iPhone.. The SimCYP Population-based ADME Simulator is a platform for the prediction of drug-drug interactions and pharmacokinetic outcomes in clinical populations. For iPhone. Distributed by SimCYP.
- Cloe Predict. Pharmacokinetic prediction using phisiologically based pharmacokinetic modeling (PBPK), and prediction of human intestinal absorption using solubility, pKa and Caco-2 permeability data. Distributed by Cyprotex Discovery.
- KnowItAll - ADME | Tox Edition. Prediction of ADME Tox properties using consensus modeling. Distributed by Bio-Rad Laboratories.
- PASS. Identification of probable targets and mechanisms of toxicity.
- MetaDrug. Predicts toxicity and metabolism of compounds using >70 QSAR models for ADME/Tox properties. Distributed by Thomson ReutersLC.
- MetaSite. Computational procedure that predicts metabolic transformations related to cytochrome-mediated reactions in phase I metabolism. The method predicts "hot spots" in the molecule, suggests the regions that contribute most towards each "hot spot", providing additional derivation sites for chemists to design new stable compounds, predicts the structures of the most likely metabolites and warns about the potential of CYP mechanism-based inhibition. Distributed by Moldiscovery.
- IMPACTS. In-silico Metabolism Prediction by A ctivated Cytochromes and Transition States (IMPACTS) predicts site of metabolism on small molecules by CYP450. This program combines docking to metabolic enzymes, transition state modeling, and rule-based substrate reactivity prediction. It is included in the Forecaser suite and provided by Molecular Forecaster Inc.
- FAME2. Program to predict site of metabolism and regioselectivity of CYP450 oxidation. Machine learnin approach relying on randomized trees and simple 2D descriptors. Software package free of charge from the Department of Computer Science, Center for Bioinformatics, Universität Hamburg, Germany.
- StarDrop. Allows the identification of the region of a molecule that are the most vulnerable to metabolism by the major drug metabolising isoforms of cytochrome P450. Distributed by optibrium.
- isoCYP. Software for the prediction of the predominant human cytochrome P450 isoform by which a given chemical compound is metabolized in phase I. Distributed by Molecular Networks.
Web services
- ALOGPS. On-line prediction of logP, water solubility and pKa(s) of compounds for drug design (ADME/T and HTS) and environmental chemistry studies. ALOGPS also displays values calculated with Pharma Algorithms LogP, LogS and pKa, Actelion LogP & LogS (many thanks to Dr Thomas Sander), Molinspiration logP, KOWWIN logP, ALOGP (Viswanadhan et al, 1989), MLOGP (Moriguchi et al, 1992) implemented in the DragonX software, XLOGP2 and XLOGP3 programs and ChemAxon logP calculator
- OSIRIS Property Explorer. Integral part of Actelion's inhouse substance registration system. Calculates on-the-fly various drug-relevant properties for drawn chemical structures, including some toxicity and druglikeness properties. Maintained by the Virtual Computational Chemistry Laboratory.
- SwissADME. A web tool that gives access to a pool of fast yet robust predictive models for physicochemical properties, pharmacokinetics, druglikeness and medicinal chemistry friendliness, among which in-house proficient methods such as the BOILED-Egg, iLOGP, Bioavailability radar and Synthetic Accessibility score. Easy efficient input and interpretation are ensured thanks to the user-friendly interface through a login-free website. Sepcialists, but also nonexperts in chemoinformatics and computational chemistry can predict rapidly key parameters for a collecion of molecules to support their medicinal chemistry endeavors. Developed and maintained by the Molecular Modeling Group of the SIB Swiss Institute of Bioinformatics.
- Chemicalize. Calculates or predict molecular properties, including logP, tautomers, PSA, pK, lipinski-like filters, etc. Provided by ChemAxon.
- AquaSol. Predicts aqueous solubility of small molecules using UG-RNN ensembles. Provided by the University of california, Irvine.
- Molinfo. Calculates or predict molecular properties other than 3D structure. Provided by the University of california, Irvine.
- ToxCreate. Web service to create computational models to predict toxicity. Provided by OpenTox.
- ADME-Tox. ADME-Tox (poor absorption, distribution, metabolism, elimination (ADME) or toxicity) filtering for small compounds, based on a set of elementary rules.
- ToxPredict. Web service to estimate toxicological hazard of a chemical structure. Molecules can be drawn, or input by any identifier (CAS, Name, EINECS) or SMILES or InChI or URL of OpenTox compound or dataset. Provided by OpenTox.
- ToxiPred. A server for prediction of aqueous toxicity of small chemical molecules in T. pyriformis. User can submit chemical molecules in the commonly used format (mol/SMILE/sdf) and after descriptors calculation the server will predict the pIGC50 value of the molecule.
- DrugMint. Web server predicting the drug-likeness of compounds.
- STITCH. Resource to explore known and predicted interactions of chemicals and proteins. Chemicals are linked to other chemicals and proteins by evidence derived from experiments, databases and the literature. STITCH contains interactions for over 74,000 small molecules and over 2.5 million proteins in 630 organisms.
- PPS. (UM-BBD Pathway Prediction System). Webservice to predict plausible pathways for microbial degradation of chemical compounds. Predictions use biotransformation rules, based on reactions found in the UM-BBD database or in the scientific literature. A list of all rules is available. Maintained by the University of Minnesota.
- DrugLogit. Web service to predict the probability of a compound being classified as a drug or non-drug, as well as disease category (or organ) classification (DC). Maintained by the Institute of Chemistry, University of Tartu, Estonia.
- XScore-LogP. Calculates the octanol/water partition coefficient for a drug, based on a feature of the X-Score program.
- VirtualToxLab. ''In silico'' tool for predicting the toxic (endocrine-disrupting) potential of existing and hypothetical compounds (drugs, chemicals, natural products) by simulating and quantifying their interactions towards a series of proteins known to trigger adverse effects using automated, flexible docking combined with multi-dimensional QSAR (mQSAR).
- pkCSM. A novel approach to the prediction of pharmacokinetic properties, which relies on graph-based signatures. These encode distance patterns between atoms and are used to represent the small molecule and to train predictive models. They were successfully used across five main different pharmacokinetic properties classes to develop predictive regression and classification models. A web server to provide an integrated freely available platform to rapidly screen multiple pharmacokinetic properties was developed by the University of Cambridge, UK.
- admetSAR. admetSAR provides the manually curated data for diverse chemicals associated with known Absorption, Distribution, Metabolism, Excretion and Toxicity profiles. admetSAR allows searching for ADMET properties profiling by name, CASRN and similarity search. In addition, admetSAR can predict about 50 ADMET endpoints by our recently development chemoinformatics-based toolbox, entitled ADMET-Simulator.
- Pred-Skin. Pred-Skin is based on statistically significant and externally predictive QSAR models of skin sensitization. The models were built using a large database containing human and murine local lymph node assay data. Developed and provided freely for diverse plateforms by the Laboratory for Molecular Modeling and Drug Design, Federal University of Goiás, Brazil.
- Pred-hERG. Pred-hERG is based on statistically significant and externally predictive QSAR models. The models were built using 16,932 entry in ChEMBL associated with bioactivity on hERG. Developed and provided freely for diverse plateforms by the Laboratory for Molecular Modeling and Drug Design, Federal University of Goiás, Brazil.
- PharmMapper. Freely accessed web-server designed to identify potential target candidates for the given probe small molecules (drugs, natural products, or other newly discovered compounds with binding targets unidentified) using pharmacophore mapping approach.
- MODEL - Molecular Descriptor Lab. Computes structural and physichemical properties of molecules from their 3D structures. Maintained by the University of Singapore.
- PreADMET. Web-based application for predicting ADME data and building drug-like library using in silico method.
- Free ADME Tools. ADME Prediction Toolbox of the SimCYP application provided free of charge by SimCYP.
- Lazar. Lazy Structure Activity Relationships. Derives predictions from toxicity databases by searching for similar compounds. provided free of charge by in silico toxicology.
- UM-BBD Pathway Prediction System. The PPS predicts plausible pathways for microbial degradation of chemical compounds. Predictions use biotransformation rules, based on reactions found in the UM-BBD database or in the scientific literature. Provided by the University of Minnesota.
- MetaPrint2D. Metabolic site predictor. MetaPrint2D is a tool that predicts xenobiotic metabolism through data-mining and statistical analysis of known metabolic transformations reported in scientific literature. MetaPrint2D-React can make predictions concerning a wider range of reactions than MetaPrint2D, and is able to predict the types of transformation that can take place at each site of metabolism, and the likely metabolite formed. Provided by the University of Cambridge.
- RA. Way2drug RA is a web-service for in silico prediction of reacting atoms. Prediction of sites of transformation for drug-like compounds for nine classes of metabolic reactions. Provided by the Institute of Biomedical Chemistry, Moscow, Russia
- MetaPrint2D-React.. Metabolic site predictor. MetaPrint2D is a tool that predicts xenobiotic metabolism through data-mining and statistical analysis of known metabolic transformations reported in scientific literature. MetaPrint2D, which predicts sites of phase I metabolism, defined as the addition of oxygen (e.g. hydroxylation, oxidation, epoxidation) or elimination reactions. Provided by the University of Cambridge.
- MetaTox. MetaTox uses a collection of biotransformation reactions and QSAR models to predict the structure, probability and toxicity of metabolites for a given input molecules. Provided as a free web service by the Institute of Biomedical Chemistry (IBMC), Moscow, Russia.
- MetaPred. MetaPred Server predict metabolizing CYP isoform of a drug molecule/substrate, based on SVM models developed using CDK descriptors.
- Property calculator. Create a physicochemical property profile for a compound. Provided by mcule.
- Aggregator Advisor. Free web service to suggest molecules that aggregate or may aggregate under biochemical assay conditions. The approach is based on the chemical similarity to known aggregators, and physical properties. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Toxicity checker. Webserver for searching substructures commonly found in toxic and promiscuous ligands. Based on more than 100 SMARTS toxic matching rules. Provided by mcule.
Databases
- PACT-F. (Preclinical And Clinical Trials Knowledge Base on Bioavailability). Preclinical And Clinical Trials Knowledge Base on Bioavailability (PACT-F). The database contains 8296 records, which describe in detail the results of clinical trials in humans and preclinical trials in animals. PACT-F is extensively annotated. Up to 17 fields describe in detail the results and conditions of each trial, such as route of administration, species investigated, drug formulation, coadministration of drug, feeding condition, age and gender of the subjects involved, dosing scheme, genetic differences, experimental and analytical procedure, method of calculation and state of health. Provided by PharmaInformatic, Germany.
- TOXNET. Databases on toxicology, hazardous chemicals, environmental health, and toxic releases that can be accessed using a common search interface. provided by the Unied States NLM.
- Leadscope Toxicity Database. Database of 160,000 chemical structures with toxicity data. Distributed by Leadscope.
- WOMBAT-PK. Database for Clinical Pharmacokinetics and Drug Target Information. WOMBAT-PK contains 1260 entries (1260 unique SMILES), totaling over 9,450 clinical pharmacokinetic measurements; it further includes 2,316 physico-chemical properties; 932 toxicity endpoints, and 2,186 annotated drug-target bioactivities. Compiled by Sunset Molecular Discovery LLC.
- Cloe Knowledge. Open Access ADME/PK Database for a range of marketed drugs. Maintained by Cyprotex.
- PHYSPROP. The Physical Properties Database (PHYSPROP) contains chemical structures, names, and physical properties for over 41,000 chemicals. Physical properties are collected from a wide variety of sources, and include experimental, extrapolated, and estimated values for melting point, boiling point, water solubility, octanol-water partition coefficient, vapor pressure, pKa, Henry's Law Constant, and OH rate constant in the atmosphere. Maintained by SRC.
- SIDER. Contains information on marketed medicines and their recorded adverse drug reactions. The information is extracted from public documents and package inserts. The available information include side effect frequency, drug and side effect classifications as well as links to further information, for example drug–target relations. Provided by the European Molecular Biology Laboratory, Heidelberg, Germany.
- admetSAR. admetSAR provides the manually curated data for diverse chemicals associated with known Absorption, Distribution, Metabolism, Excretion and Toxicity profiles. admetSAR allows searching for ADMET properties profiling by name, CASRN and similarity search. In addition, admetSAR can predict about 50 ADMET endpoints by our recently development chemoinformatics-based toolbox, entitled ADMET-Simulator.
- The ADME databases. Databases for benchmarking the results of experiments, validating the accuracy of existing ADME predictive models, and building new predictive models.
- The ADME database. Provides comprehensive data for structurally diverse compounds associated with known ADME properties, including human oral bioavailability, enzymes metabolism, inhibition and induction, transport, plasma protein binding and bloodbrain barrier. Distributed by Aureus.
- UCSF-FDA Transportal. The purpose of this database is to be a useful repository of information on transporters important in the drug discovery process as a part of the US Food and Drug Administration-led Critical Path Initiative. Information includes transporter expression, localization, substrates, inhibitors, and drug-drug interactions It contains 3438 compounds, 11649 interaction records, 1211 literature references. The FDA has partnered with the Giacomini lab at UCSF to create a transporter database of pharmacologically relevant transporters to support development of new pharmaceuticals. Information on important transporters, their localization, expression levels, substrates, and inhibitors have been curated from the literature and compiled into a single location to aid and inform drug developers, regulatory agencies and academic scientists about transporters important in drug action and disposition.. The database will also help drug developers in determining what experiments or analyses must be conducted to check for possible drug interactions through transporters as well as identify promising transporter candidates for the testing of possible genetic influences.
- SuperTarget Database. Database of about 332828 drug-target relations.
- DART. (Drug Adverse Reaction Target). A database for facilitating the search for drug adverse reaction target. It contains information about known drug adverse rection targets, functions and properties. Associated references are also included. Maintained by the University of Singapore.
- DITOP. (Drug-Induced Toxicity Related Proteins). Database of proteins that mediate toxicities through their interaction with drugs or reactive metabolites. Can be searched using keywords of chemicals, proteins, or toxicity terms. Maintained by the Xiamen university.
- ADMEAP. A database for facilitating the search for drug Absorption, Distribution, Metabolism, Excretion associated proteins. It contains information about known drug ADME associated proteins, functions, similarities, substrates / ligands, tissue distributions, and other properties of the targets. Associated references are also included. Currently this database contains 321 protein entries. Maintained by the Dept.Computational Science. NUS.
- SIDER. (Side Effect Resource). contains information on marketed medicines and their recorded adverse drug reactions. The information is extracted from public documents and package inserts. The available information include side effect frequency, drug and side effect classifications as well as links to further information, for example drug–target relations.
- SAR Genetox Database. Genetic toxicity database to be used as a resource for developing predictive modeling training sets. Distributed by Leadscope.
- SAR Carcinogenicity Database. Carcinogenicity database with validated structures to be used as a resource for preparing training sets. Distributed by Leadscope.
- HMDB. The Human Metabolome Database (HMDB) is a freely available electronic database containing detailed information about small molecule metabolites found in the human body. The database contains chemical data, clinical data, and molecular biology/biochemistry data. The database (version 2.5) contains over 7900 metabolite entries including both water-soluble and lipid soluble metabolites as well as metabolites that would be regarded as either abundant (> 1 uM) or relatively rare (< 1 nM). Provided by the Departments of Computing Science & Biological Sciences, University of Alberta.
- t3db. (Toxin and Toxin Target Database). Combines detailed toxin data with comprehensive toxin target information. The database currently houses over 2900 toxins described by over 34 200 synonyms, including pollutants, pesticides, drugs, and food toxins, which are linked to over 1300 corresponding toxin target records. Altogether there are over 33 800 toxin, toxin target associations. Each toxin record (ToxCard) contains over 50 data fields and holds information such as chemical properties and descriptors, toxicity values, molecular and cellular interactions, and medical information. This information has been extracted from over 5600 sources, which include other databases, government documents, books, and scientific literature. Provided by the Departments of Computing Science & Biological Sciences, University of Alberta.
- SuperToxic. Collection of toxic compounds from literature and web sources. The current version of this database compiles approx. 60,000 compounds with about 100,000 synonyms. These molecules are classified according to their toxicity based on more than 2,500,000 measurements. Provided by Charité Berlin, Structural Bioinformatics Group.
- SuperHapten. Comprehensive database for small immunogenic compounds. Contains currently 7257 haptens, 453 commercially available related antibodies and 24 carriers. Provided by Charité Berlin, Institute of Molecular Biology and Bioinformatics.
- HaptenDB. Database of about 1087 haptens that includes common and chemical name of Hapten, molecular mass, physical and chemical properties, biological importance and the structure. Provided by the Institute of Microbial Technology, India.
- SuperCyp. Comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Provided by Charité Berlin, Structural Bioinformatics Group.
- PROMISCUOUS. Exhaustive resource of protein-protein and drug-protein interactions with the aim of providing a uniform data set for drug repositioning and further analysis. PROMISCUOUS contains three different types of entities: drugs, proteins and side-effects as well as relations between them. Provided by Charité Berlin, Structural Bioinformatics Group.
Contacts : Vincent.Zoete@sib.swiss or Antoine.Daina@sib.swiss